Hannon Eilis, Weedon Mike, Bray Nicholas, O'Donovan Michael, Mill Jonathan
University of Exeter Medical School, Exeter EX2 5DW, UK.
MRC Centre for Neuropsychiatric Genetics and Genomics, Cardiff University School of Medicine, Cardiff CF24 4HQ, UK.
Am J Hum Genet. 2017 Jun 1;100(6):954-959. doi: 10.1016/j.ajhg.2017.04.013. Epub 2017 May 18.
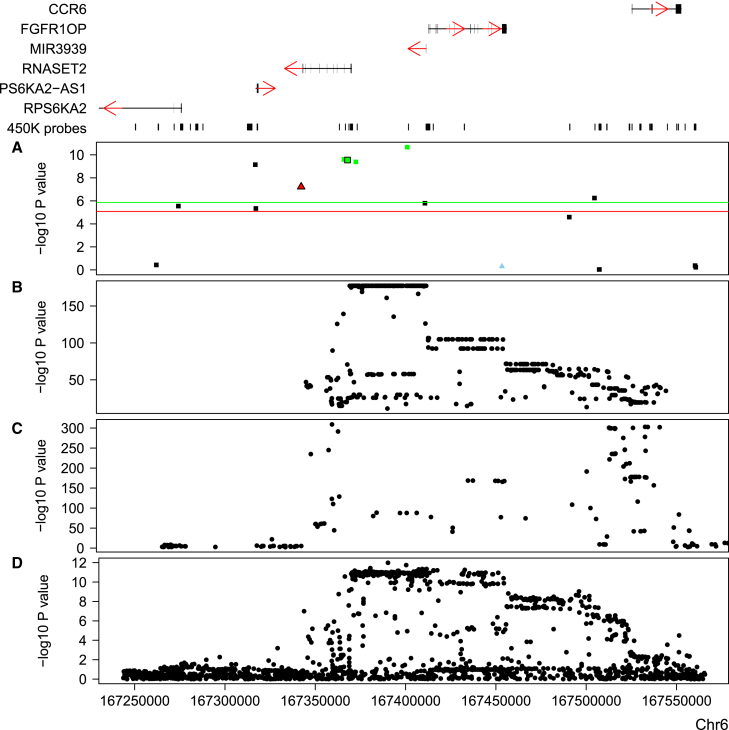
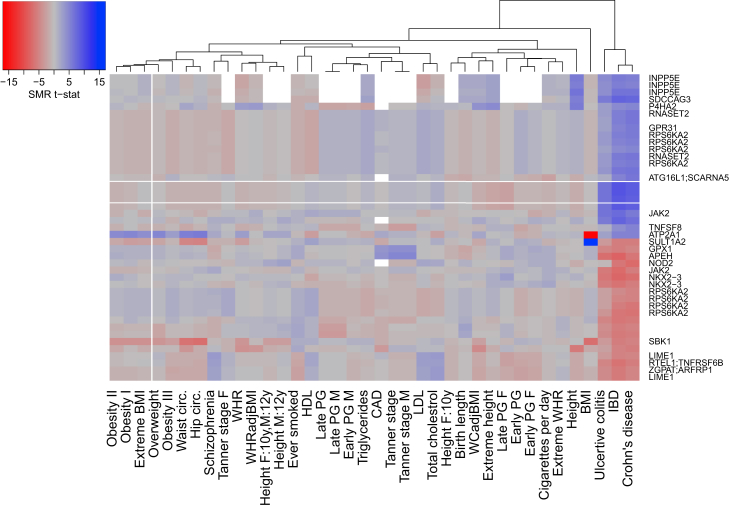
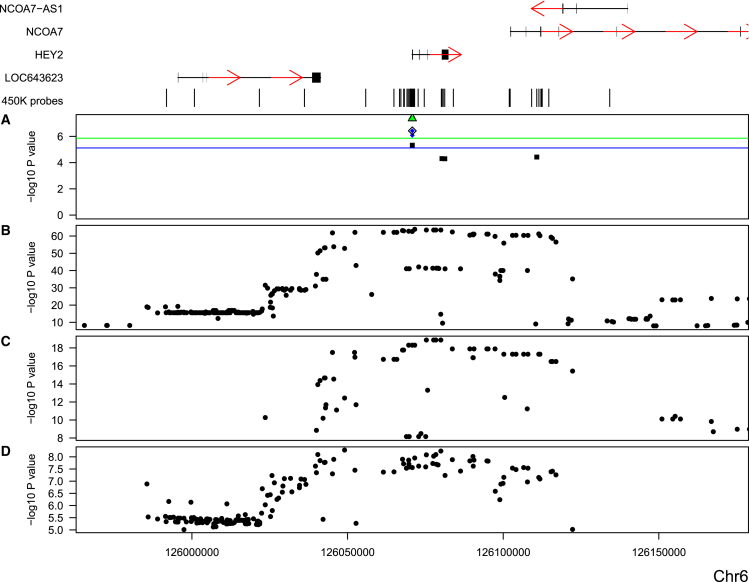
Most genetic variants identified in genome-wide association studies (GWASs) of complex traits are thought to act by affecting gene regulation rather than directly altering the protein product. As a consequence, the actual genes involved in disease are not necessarily the most proximal to the associated variants. By integrating data from GWAS analyses with those from genetic studies of regulatory variation, it is possible to identify variants pleiotropically associated with both a complex trait and measures of gene regulation. In this study, we used summary-data-based Mendelian randomization (SMR), a method developed to identify variants pleiotropically associated with both complex traits and gene expression, to identify variants associated with complex traits and DNA methylation. We used large DNA methylation quantitative trait locus (mQTL) datasets generated from two different tissues (blood and fetal brain) to prioritize genes for >40 complex traits with robust GWAS data and found considerable overlap with the results of SMR analyses performed with expression QTL (eQTL) data. We identified multiple examples of variable DNA methylation associated with GWAS variants for a range of complex traits, demonstrating the utility of this approach for refining genetic association signals.
在复杂性状的全基因组关联研究(GWAS)中鉴定出的大多数基因变异,被认为是通过影响基因调控而非直接改变蛋白质产物来发挥作用的。因此,涉及疾病的实际基因不一定是与相关变异最接近的基因。通过将GWAS分析数据与调控变异的遗传学研究数据相结合,有可能识别出与复杂性状和基因调控指标多效性相关的变异。在本研究中,我们使用基于汇总数据的孟德尔随机化(SMR)方法(一种为识别与复杂性状和基因表达多效性相关的变异而开发的方法)来识别与复杂性状和DNA甲基化相关的变异。我们使用从两种不同组织(血液和胎儿大脑)生成的大型DNA甲基化定量性状位点(mQTL)数据集,对具有可靠GWAS数据的40多种复杂性状的基因进行优先级排序,并发现与使用表达QTL(eQTL)数据进行的SMR分析结果有相当大的重叠。我们确定了一系列复杂性状的与GWAS变异相关的可变DNA甲基化的多个实例,证明了这种方法在细化遗传关联信号方面的实用性。