Li Xihua, Wu Man, Liu Guoyuan, Pei Wenfeng, Zhai Honghong, Yu Jiwen, Zhang Jinfa, Yu Shuxun
College of Agronomy, Northwest A&F University, Yangling, 712100, China.
State Key Laboratory of Cotton Biology, Institute of Cotton Research, Chinese Academy of Agricultural Science, Anyang, 455000, China.
BMC Genomics. 2017 May 31;18(1):427. doi: 10.1186/s12864-017-3812-5.
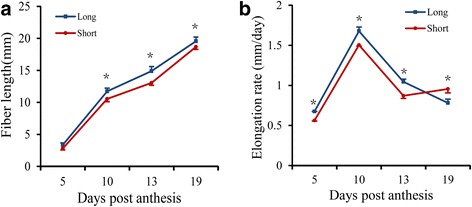
Cotton (Gossypium spp.) fibers are single-celled elongated trichomes, the molecular aspects of genetic variation in fiber length (FL) among genotypes are currently unknown. In this study, two backcross inbred lines (BILs), i.e., NMGA-062 ("Long") and NMGA-105 ("Short") with 32.1 vs. 27.2 mm in FL, respectively, were chosen to perform RNA-Seq on developing fibers at 10 days post anthesis (DPA). The two BILs differed in 4 quantitative trait loci (QTL) for FL and were developed from backcrosses between G. hirsutum as the recurrent parent and G. barbadense.
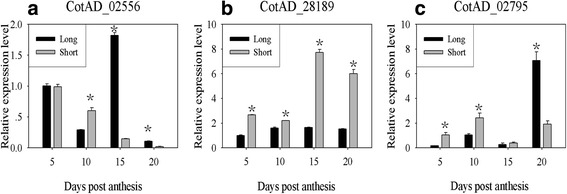
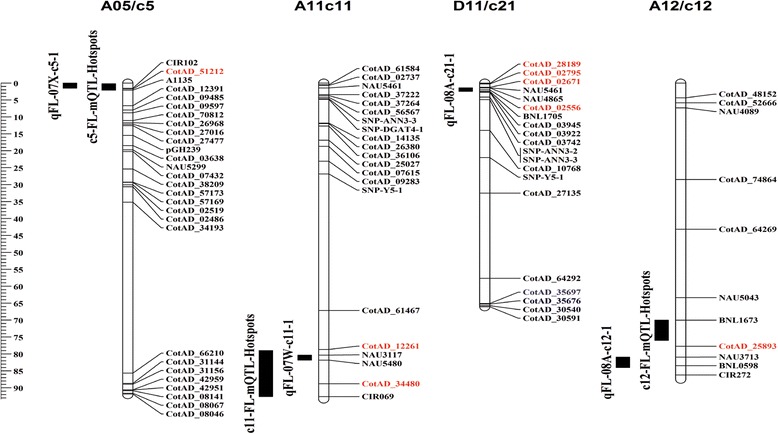
In total, 51.7 and 54.3 million reads were obtained and assembled to 49,508 and 49,448 transcripts in the two genotypes, respectively. Of 1551 differentially expressed genes (DEGs) between the two BILs, 678 were up-regulated and 873 down-regulated in "Long"; and 703 SNPs were identified in 339 DEGs. Further physical mapping showed that 8 DEGs were co-localized with the 4 FL QTL identified in the BIL population containing the two BILs. Four SNP markers in 3 DEGs that showed significant correlations with FL were developed. Among the three candidate genes encoding for proline-rich protein, D-cysteine desulfhydrase, and thaumatin-like protein, a SNP of thaumatin-like protein gene showed consistent correlations with FL across all testing environments.
This study represents one of the first investigations of positional candidate gene approach of QTL in cotton in integrating transcriptome and SNP identification based on RNA-Seq with linkage and physical mapping of QTL and genes, which will facilitate eventual cloning and identification of genes responsible for FL QTL. The candidate genes may serve as the foundation for further in-depth studies of the molecular mechanism of natural variation in fiber elongation.
棉花(棉属)纤维是单细胞伸长的表皮毛,目前不同基因型间纤维长度(FL)遗传变异的分子机制尚不清楚。本研究选择了两个回交自交系(BILs),即NMGA - 062(“长纤维”)和NMGA - 105(“短纤维”),其纤维长度分别为32.1毫米和27.2毫米,在开花后10天(DPA)对发育中的纤维进行RNA测序。这两个BILs在纤维长度的4个数量性状位点(QTL)上存在差异,它们是由陆地棉作为轮回亲本与海岛棉回交培育而成。
两个基因型分别获得了总计5170万和5430万条 reads,并组装成49508和49448个转录本。在两个BILs之间的1551个差异表达基因(DEGs)中,“长纤维”中有678个上调,873个下调;在339个DEGs中鉴定出703个单核苷酸多态性(SNPs)。进一步的物理定位表明,8个DEGs与在包含这两个BILs的BIL群体中鉴定出的4个纤维长度QTL共定位。在3个与纤维长度显著相关的DEGs中开发了4个SNP标记。在编码富含脯氨酸蛋白、D - 半胱氨酸脱硫酶和类甜蛋白的三个候选基因中,类甜蛋白基因的一个SNP在所有测试环境中与纤维长度均表现出一致的相关性。
本研究首次基于RNA测序将转录组和SNP鉴定与QTL和基因的连锁及物理定位相结合,对棉花QTL的位置候选基因方法进行研究,这将有助于最终克隆和鉴定负责纤维长度QTL的基因。这些候选基因可为进一步深入研究纤维伸长自然变异的分子机制奠定基础。