Chelban Viorica, Patel Nisha, Vandrovcova Jana, Zanetti M Natalia, Lynch David S, Ryten Mina, Botía Juan A, Bello Oscar, Tribollet Eloise, Efthymiou Stephanie, Davagnanam Indran, Bashiri Fahad A, Wood Nicholas W, Rothman James E, Alkuraya Fowzan S, Houlden Henry
Department of Molecular Neuroscience, University College London, London WC1N 3BG, UK; Department of Neurology and Neurosurgery, Institute of Emergency Medicine, Toma Ciorbă 1, 2052 Chisinau, Republic of Moldova.
Developmental Genetics Unit, Department of Genetics, King Faisal Specialist Hospital and Research Center, MBC 03, PO Box 3354, Riyadh 11211 Saudi Arabia.
Am J Hum Genet. 2017 Jun 1;100(6):969-977. doi: 10.1016/j.ajhg.2017.05.009.
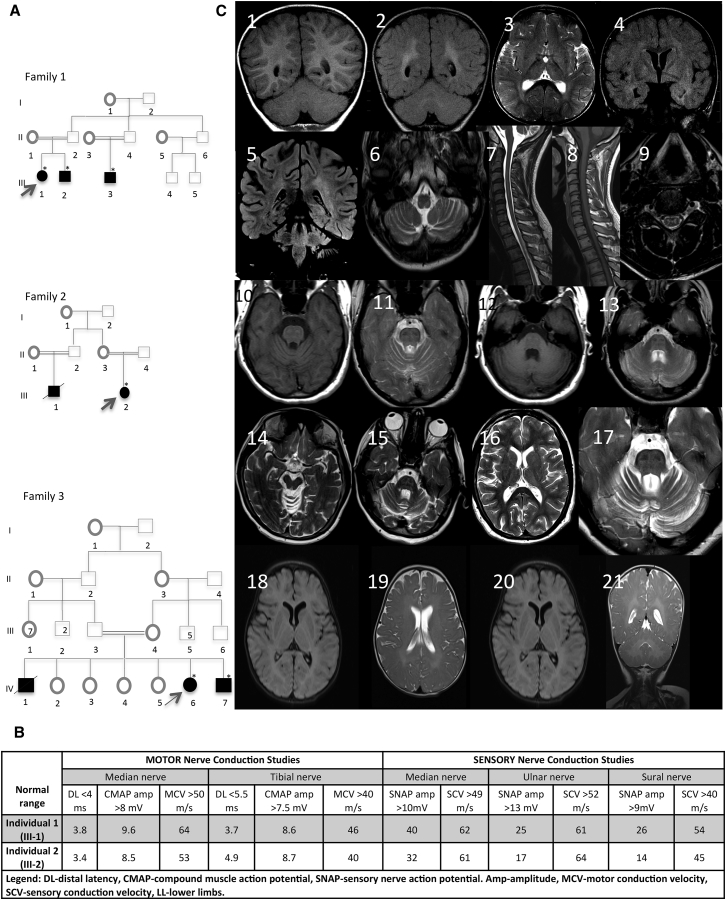
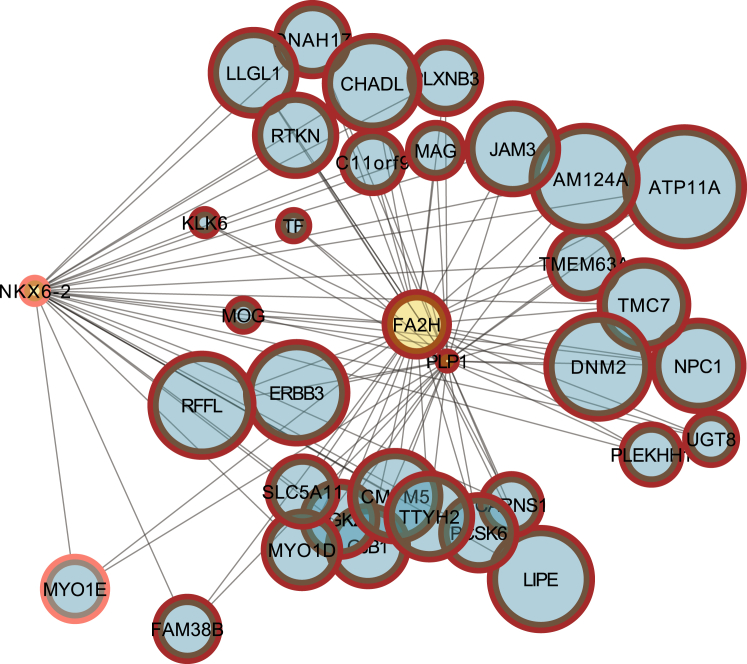
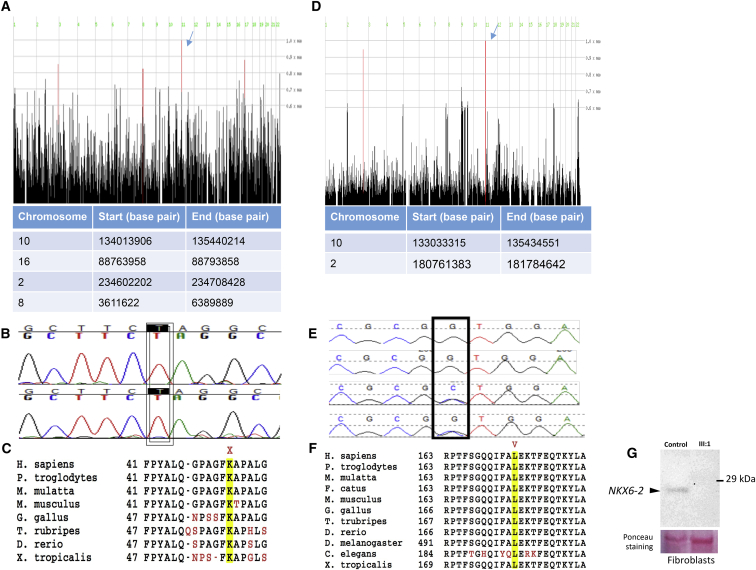
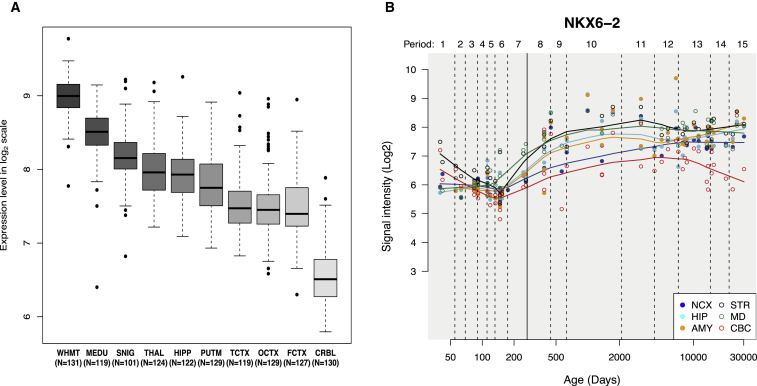
Progressive limb spasticity and cerebellar ataxia are frequently found together in clinical practice and form a heterogeneous group of degenerative disorders that are classified either as pure spastic ataxia or as complex spastic ataxia with additional neurological signs. Inheritance is either autosomal dominant or autosomal recessive. Hypomyelinating features on MRI are sometimes seen with spastic ataxia, but this is usually mild in adults and severe and life limiting in children. We report seven individuals with an early-onset spastic-ataxia phenotype. The individuals come from three families of different ethnic backgrounds. Affected members of two families had childhood onset disease with very slow progression. They are still alive in their 30s and 40s and show predominant ataxia and cerebellar atrophy features on imaging. Affected members of the third family had a similar but earlier-onset presentation associated with brain hypomyelination. Using a combination of homozygozity mapping and exome sequencing, we mapped this phenotype to deleterious nonsense or homeobox domain missense mutations in NKX6-2. NKX6-2 encodes a transcriptional repressor with early high general and late focused CNS expression. Deficiency of its mouse ortholog results in widespread hypomyelination in the brain and optic nerve, as well as in poor motor coordination in a pattern consistent with the observed human phenotype. In-silico analysis of human brain expression and network data provides evidence that NKX6-2 is involved in oligodendrocyte maturation and might act within the same pathways of genes already associated with central hypomyelination. Our results support a non-redundant developmental role of NKX6-2 in humans and imply that NKX6-2 mutations should be considered in the differential diagnosis of spastic ataxia and hypomyelination.
进行性肢体痉挛和小脑共济失调在临床实践中经常同时出现,形成一组异质性的退行性疾病,可分为单纯痉挛性共济失调或伴有其他神经体征的复杂痉挛性共济失调。遗传方式为常染色体显性或常染色体隐性。MRI上的低髓鞘形成特征有时见于痉挛性共济失调,但在成人中通常较轻,在儿童中则严重且危及生命。我们报告了7例具有早发性痉挛性共济失调表型的个体。这些个体来自三个不同种族背景的家庭。两个家庭的受影响成员在儿童期发病,病情进展非常缓慢。他们在30多岁和40多岁时仍然活着,影像学上显示主要为共济失调和小脑萎缩特征。第三个家庭的受影响成员有类似但更早发病的表现,并伴有脑低髓鞘形成。通过纯合子定位和外显子组测序相结合的方法,我们将这种表型定位到NKX6-2基因的有害无义突变或同源盒结构域错义突变。NKX6-2编码一种转录抑制因子,在中枢神经系统早期广泛表达,后期表达集中。其小鼠同源基因的缺陷导致大脑和视神经广泛的低髓鞘形成,以及运动协调性差,其模式与观察到的人类表型一致。对人类大脑表达和网络数据的电子分析提供了证据,表明NKX6-2参与少突胶质细胞成熟,可能在已经与中枢低髓鞘形成相关的基因的相同途径中起作用。我们的结果支持NKX6-2在人类发育中具有非冗余作用,并暗示在痉挛性共济失调和低髓鞘形成的鉴别诊断中应考虑NKX6-2突变。