Hamilton Eline M C, Bertini Enrico, Kalaydjieva Luba, Morar Bharti, Dojčáková Dana, Liu Judy, Vanderver Adeline, Curiel Julian, Persoon Claudia M, Diodato Daria, Pinelli Lorenzo, van der Meij Nathalie L, Plecko Barbara, Blaser Susan, Wolf Nicole I, Waisfisz Quinten, Abbink Truus E M, van der Knaap Marjo S
From the Department of Child Neurology (E.M.C.H., N.I.W., T.E.M.A., M.S.v.d.K.), Amsterdam Neuroscience (E.M.C.H., N.I.W., T.E.M.A., M.S.v.d.K.), Department of Clinical Genetics (C.M.P., Q.W.), Department of Functional Genomics, Center for Neurogenomics and Cognitive Research (M.S.v.d.K.), VU University and VU University Medical Center, Amsterdam, the Netherlands; Unit of Neuromuscular and Neurodegenerative Disorders (E.B., D. Diodato), Laboratory of Molecular Medicine, "Bambino Gesù" Children's Hospital, IRCCS, Rome, Italy; Harry Perkins Institute of Medical Research and Centre for Medical Research (L.K., B.M.), University of Western Australia, Perth; Department of Biology (D. Dojčáková), Faculty of Humanities and Natural Sciences, University of Presov, Slovakia; Center for Neuroscience Research (J.L., J.C.), Children's Research Institute; Department of Neurology, Center for Genetic Medicine Research (A.V.), Children's National Medical Center, Washington, DC; Department of Neuroradiology (L.P.), Section of Pediatric Neuroradiology, Spedali Civili, Brescia, Italy; MRC Holland (N.L.v.d.M.), Amsterdam, the Netherlands; Division of Neurology (B.P.), Children's Hospital, University of Zurich, Switzerland; and Division of Pediatric Neuroradiology (S.B.), Hospital for Sick Children, Toronto, Canada.
Neurology. 2017 Oct 24;89(17):1821-1828. doi: 10.1212/WNL.0000000000004578. Epub 2017 Sep 20.
To identify the gene defect in patients with hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) who are negative for mutations.
We performed homozygosity mapping and whole exome sequencing (WES) to detect the disease-causing variant. We used a Taqman assay for population screening. We developed a luciferase reporter construct to investigate the effect of the promoter mutation on expression.
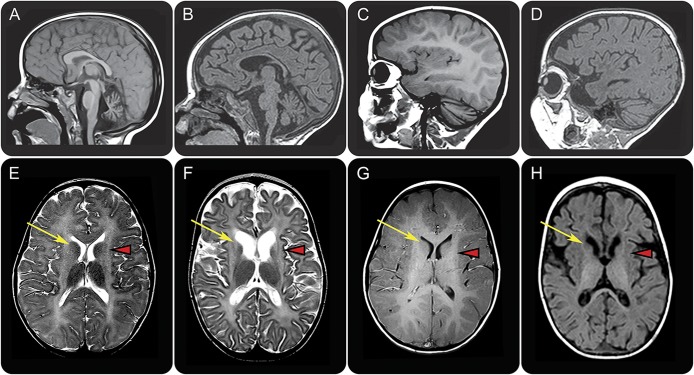
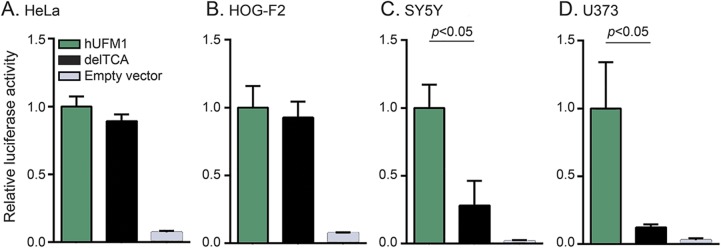
Sixteen patients from 14 families from different countries fulfilling the MRI criteria for H-ABC exhibited a similar, severe clinical phenotype, including lack of development and a severe epileptic encephalopathy. The majority of patients had a known Roma ethnic background. Single nucleotide polymorphism array analysis in 5 patients identified one large overlapping homozygous region on chromosome 13. WES in 2 patients revealed a homozygous deletion in the promoter region of . Sanger sequencing confirmed homozygosity for this variant in all 16 patients. All patients shared a common haplotype, indicative of a founder effect. Screening of 1,000 controls from different European Roma panels demonstrated an overall carrier rate of the mutation of 3%-25%. Transfection assays showed that the deletion significantly reduced expression in specific CNS cell lines.
encodes ubiquitin-fold modifier 1 (UFM1), a member of the ubiquitin-like family involved in posttranslational modification of proteins. Its exact biological role is unclear. This study associates a gene defect with a disease and sheds new light on possible UFM1 functional networks.
在基底神经节和小脑萎缩性低髓鞘化(H-ABC)且突变检测呈阴性的患者中鉴定基因缺陷。
我们进行了纯合性定位和全外显子组测序(WES)以检测致病变异。我们使用Taqman分析进行群体筛查。我们构建了荧光素酶报告基因载体以研究启动子突变对表达的影响。
来自不同国家的14个家庭的16例符合H-ABC的MRI标准的患者表现出相似的严重临床表型,包括发育迟缓以及严重的癫痫性脑病。大多数患者有已知的罗姆族裔背景。对5例患者进行的单核苷酸多态性阵列分析在13号染色体上鉴定出一个大的重叠纯合区域。对2例患者进行的WES显示在 基因的启动子区域存在纯合缺失。Sanger测序证实所有16例患者中该变异均为纯合状态。所有患者共享一个共同的单倍型,提示存在奠基者效应。对来自不同欧洲罗姆人群体的1000名对照进行筛查,结果显示该突变的总体携带率为3%-25%。转染实验表明该缺失显著降低了特定中枢神经系统细胞系中的表达。
编码泛素折叠修饰因子1(UFM1),它是泛素样家族的成员,参与蛋白质的翻译后修饰。其确切的生物学作用尚不清楚。本研究将 基因缺陷与一种疾病相关联,并为可能的UFM1功能网络提供了新的线索。