Schuierer Sven, Carbone Walter, Knehr Judith, Petitjean Virginie, Fernandez Anita, Sultan Marc, Roma Guglielmo
Novartis Institutes for Biomedical Research, Novartis Pharma AG, Basel, Switzerland.
BMC Genomics. 2017 Jun 5;18(1):442. doi: 10.1186/s12864-017-3827-y.
RNA-sequencing (RNA-seq) has emerged as one of the most sensitive tool for gene expression analysis. Among the library preparation methods available, the standard poly(A) + enrichment provides a comprehensive, detailed, and accurate view of polyadenylated RNAs. However, on samples of suboptimal quality ribosomal RNA depletion and exon capture methods have recently been reported as better alternatives.
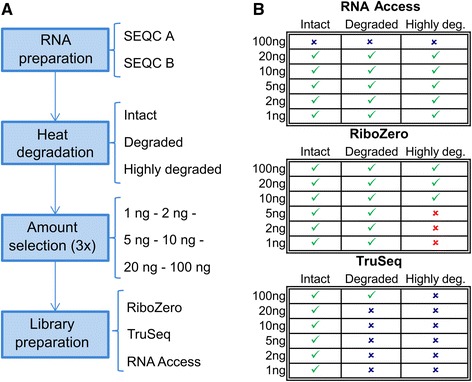
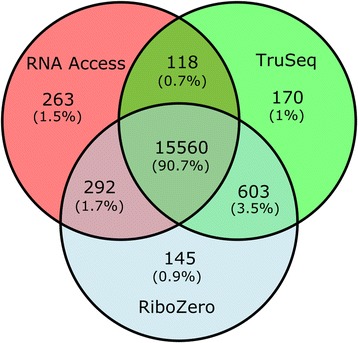
We compared for the first time three commercial Illumina library preparation kits (TruSeq Stranded mRNA, TruSeq Ribo-Zero rRNA Removal, and TruSeq RNA Access) as representatives of these three different approaches using well-established human reference RNA samples from the MAQC/SEQC consortium on a wide range of input amounts (from 100 ng down to 1 ng) and degradation levels (intact, degraded, and highly degraded).
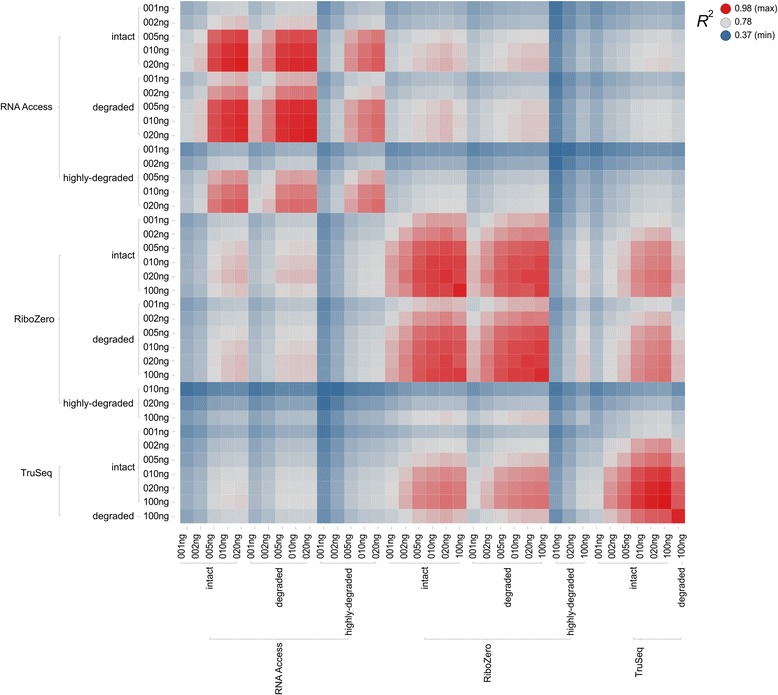
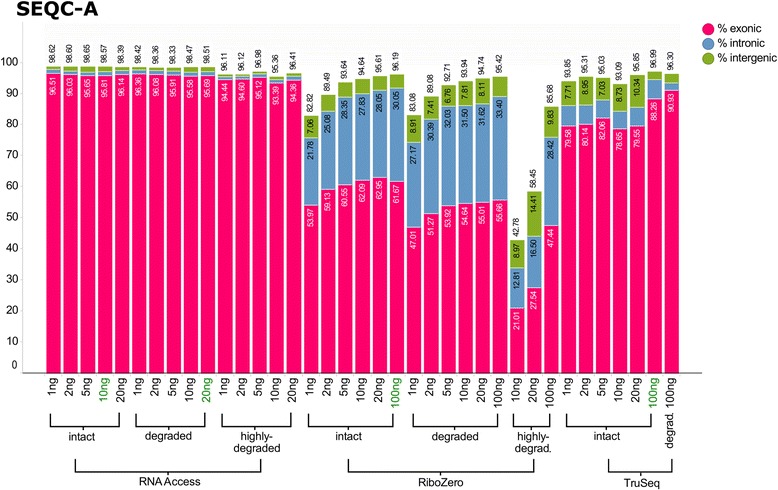
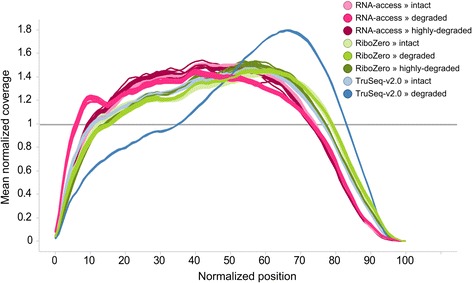
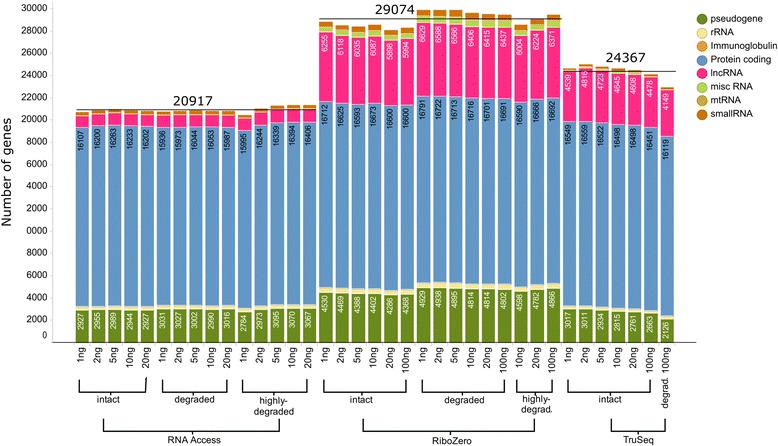
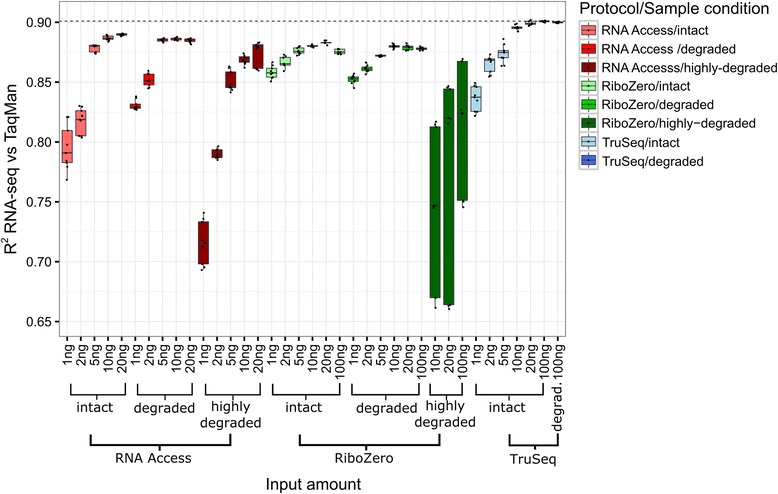
We assessed the accuracy of the generated expression values by comparison to gold standard TaqMan qPCR measurements and gained unprecedented insight into the limits of applicability in terms of input quantity and sample quality of each protocol. We found that each protocol generates highly reproducible results (R > 0.92) on intact RNA samples down to input amounts of 10 ng. For degraded RNA samples, Ribo-Zero showed clear performance advantages over the other two protocols as it generated more accurate and better reproducible gene expression results even at very low input amounts such as 1 ng and 2 ng. For highly degraded RNA samples, RNA Access performed best generating reliable data down to 5 ng input.
We found that the ribosomal RNA depletion protocol from Illumina works very well at amounts far below recommendation and over a good range of intact and degraded material. We also infer that the exome-capture protocol (RNA Access, Illumina) performs better than other methods on highly degraded and low amount samples.
RNA测序(RNA-seq)已成为基因表达分析最灵敏的工具之一。在现有的文库制备方法中,标准的聚腺苷酸(poly(A))+富集法能提供多聚腺苷酸化RNA全面、详细且准确的图谱。然而,对于质量欠佳的样本,最近有报道称核糖体RNA去除法和外显子捕获法是更好的选择。
我们首次比较了三种商业化的Illumina文库制备试剂盒(TruSeq链特异性mRNA试剂盒、TruSeq Ribo-Zero核糖体RNA去除试剂盒和TruSeq RNA Access试剂盒),它们分别代表这三种不同的方法,使用来自MAQC/SEQC联盟的成熟人类参考RNA样本,涵盖广泛的输入量(从100 ng到1 ng)和降解水平(完整、降解和高度降解)。
我们通过与金标准TaqMan定量PCR测量结果进行比较,评估了生成的表达值的准确性,并对每个方案在输入量和样本质量方面的适用范围极限有了前所未有的深入了解。我们发现,对于完整RNA样本,每种方案在低至10 ng的输入量时都能产生高度可重复的结果(R>0.92)。对于降解的RNA样本,Ribo-Zero试剂盒比其他两种方案表现出明显的性能优势,因为即使在非常低的输入量(如1 ng和2 ng)下,它也能产生更准确、可重复性更好的基因表达结果。对于高度降解的RNA样本,RNA Access试剂盒表现最佳,在低至5 ng的输入量时仍能生成可靠的数据。
我们发现Illumina的核糖体RNA去除方案在远低于推荐量的情况下,对于完整和降解材料的良好范围内都能很好地工作。我们还推断,外显子捕获方案(RNA Access,Illumina)在高度降解和低量样本上比其他方法表现更好。