Kinoti Wycliff M, Constable Fiona E, Nancarrow Narelle, Plummer Kim M, Rodoni Brendan
Agriculture Victoria, AgriBio, La Trobe University, Melbourne, VIC, Australia.
School of Applied Systems Biology, AgriBio, La Trobe University, Melbourne, VIC, Australia.
PLoS One. 2017 Jun 20;12(6):e0179284. doi: 10.1371/journal.pone.0179284. eCollection 2017.
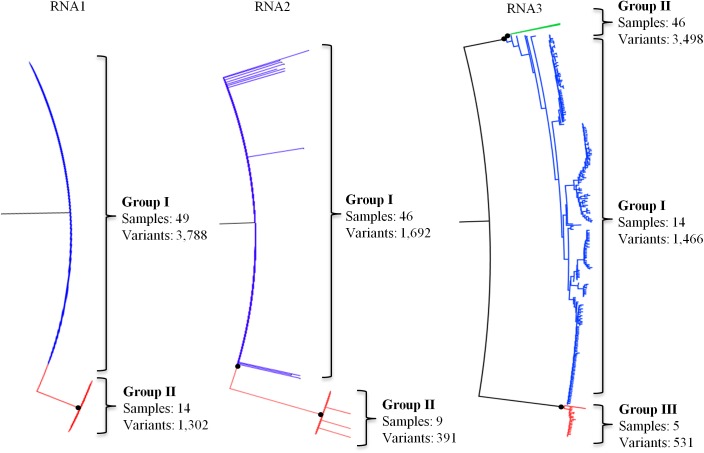
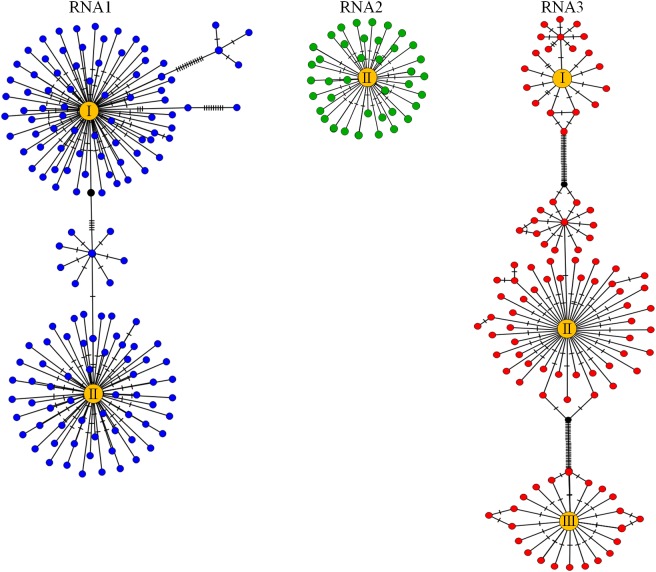
PCR amplicon next generation sequencing (NGS) analysis offers a broadly applicable and targeted approach to detect populations of both high- or low-frequency virus variants in one or more plant samples. In this study, amplicon NGS was used to explore the diversity of the tripartite genome virus, Prunus necrotic ringspot virus (PNRSV) from 53 PNRSV-infected trees using amplicons from conserved gene regions of each of PNRSV RNA1, RNA2 and RNA3. Sequencing of the amplicons from 53 PNRSV-infected trees revealed differing levels of polymorphism across the three different components of the PNRSV genome with a total number of 5040, 2083 and 5486 sequence variants observed for RNA1, RNA2 and RNA3 respectively. The RNA2 had the lowest diversity of sequences compared to RNA1 and RNA3, reflecting the lack of flexibility tolerated by the replicase gene that is encoded by this RNA component. Distinct PNRSV phylo-groups, consisting of closely related clusters of sequence variants, were observed in each of PNRSV RNA1, RNA2 and RNA3. Most plant samples had a single phylo-group for each RNA component. Haplotype network analysis showed that smaller clusters of PNRSV sequence variants were genetically connected to the largest sequence variant cluster within a phylo-group of each RNA component. Some plant samples had sequence variants occurring in multiple PNRSV phylo-groups in at least one of each RNA and these phylo-groups formed distinct clades that represent PNRSV genetic strains. Variants within the same phylo-group of each Prunus plant sample had ≥97% similarity and phylo-groups within a Prunus plant sample and between samples had less ≤97% similarity. Based on the analysis of diversity, a definition of a PNRSV genetic strain was proposed. The proposed definition was applied to determine the number of PNRSV genetic strains in each of the plant samples and the complexity in defining genetic strains in multipartite genome viruses was explored.
聚合酶链式反应(PCR)扩增子下一代测序(NGS)分析提供了一种广泛适用的靶向方法,用于检测一个或多个植物样本中高频或低频病毒变体群体。在本研究中,扩增子NGS被用于探索来自53株感染李坏死环斑病毒(PNRSV)的树木的三方基因组病毒的多样性,使用来自PNRSV RNA1、RNA2和RNA3每个保守基因区域的扩增子。对53株感染PNRSV的树木的扩增子进行测序,结果显示PNRSV基因组的三个不同组分存在不同程度的多态性,RNA1、RNA2和RNA3分别观察到5040、2083和5486个序列变体。与RNA1和RNA3相比,RNA2的序列多样性最低,这反映了该RNA组分编码的复制酶基因缺乏耐受性。在PNRSV RNA1、RNA2和RNA3中均观察到由密切相关的序列变体簇组成的不同PNRSV系统发育组。大多数植物样本的每个RNA组分都有一个单一的系统发育组。单倍型网络分析表明,PNRSV序列变体的较小簇在每个RNA组分的系统发育组内与最大的序列变体簇存在遗传联系。一些植物样本在每个RNA的至少一个中存在多个PNRSV系统发育组中的序列变体,并且这些系统发育组形成了代表PNRSV遗传株系的不同进化枝。每个李属植物样本同一系统发育组内的变体具有≥97%的相似性,而李属植物样本内和样本间的系统发育组具有≤97%的相似性。基于多样性分析,提出了PNRSV遗传株系的定义。将所提出的定义应用于确定每个植物样本中PNRSV遗传株系的数量,并探讨了在多分体基因组病毒中定义遗传株系的复杂性。