Battenberg Kai, Lee Ernest K, Chiu Joanna C, Berry Alison M, Potter Daniel
Department of Plant Sciences, University of California, Davis, CA, USA.
Department of Entomology and Nematology, University of California, Davis, CA, USA.
BMC Bioinformatics. 2017 Jun 21;18(1):310. doi: 10.1186/s12859-017-1726-5.
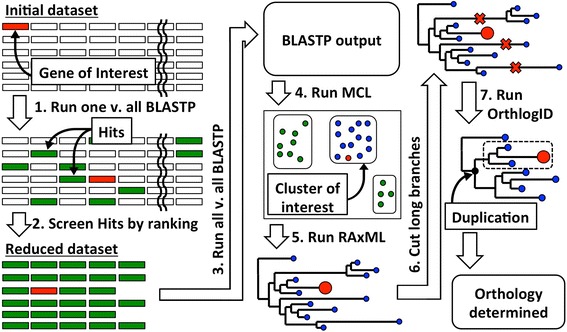
Identifying orthologous genes is an initial step required for phylogenetics, and it is also a common strategy employed in functional genetics to find candidates for functionally equivalent genes across multiple species. At the same time, in silico orthology prediction tools often require large computational resources only available on computing clusters. Here we present OrthoReD, an open-source orthology prediction tool with accuracy comparable to published tools that requires only a desktop computer. The low computational resource requirement of OrthoReD is achieved by repeating orthology searches on one gene of interest at a time, thereby generating a reduced dataset to limit the scope of orthology search for each gene of interest.
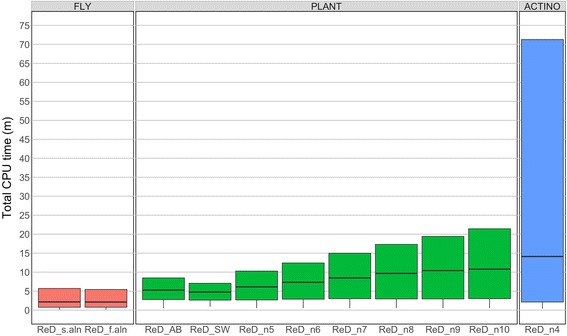
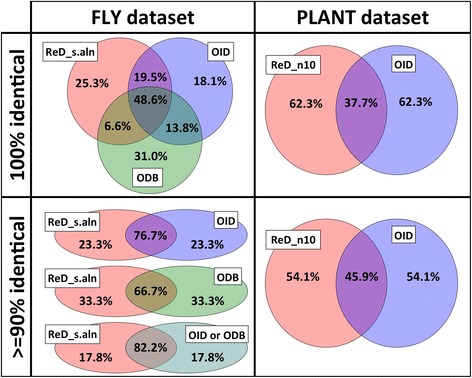
The output of OrthoReD was highly similar to the outputs of two other published orthology prediction tools, OrthologID and/or OrthoDB, for the three dataset tested, which represented three phyla with different ranges of species diversity and different number of genomes included. Median CPU time for ortholog prediction per gene by OrthoReD executed on a desktop computer was <15 min even for the largest dataset tested, which included all coding sequences of 100 bacterial species.
With high-throughput sequencing, unprecedented numbers of genes from non-model organisms are available with increasing need for clear information about their orthologies and/or functional equivalents in model organisms. OrthoReD is not only fast and accurate as an orthology prediction tool, but also gives researchers flexibility in the number of genes analyzed at a time, without requiring a high-performance computing cluster.
识别直系同源基因是系统发育学所需的第一步,也是功能遗传学中常用的策略,用于在多个物种中寻找功能等效基因的候选者。同时,计算机模拟直系同源预测工具通常需要大型计算资源,而这些资源通常只有在计算集群上才能获得。在此,我们展示了OrthoReD,这是一种开源的直系同源预测工具,其准确性与已发表的工具相当,且只需要一台台式计算机。OrthoReD对计算资源的低要求是通过一次对一个感兴趣的基因重复进行直系同源搜索来实现的,从而生成一个简化的数据集,以限制对每个感兴趣基因的直系同源搜索范围。
对于所测试的三个数据集,OrthoReD的输出与另外两个已发表的直系同源预测工具(OrthologID和/或OrthoDB)的输出高度相似,这三个数据集代表了具有不同物种多样性范围和不同基因组数量的三个门。即使对于测试的最大数据集,即在台式计算机上运行的OrthoReD预测每个基因直系同源的中位数CPU时间也<15分钟,该最大数据集包含100种细菌物种的所有编码序列。
随着高通量测序技术的发展,来自非模式生物的基因数量空前增加,人们越来越需要了解它们在模式生物中的直系同源基因和/或功能等效基因的清晰信息。OrthoReD作为一种直系同源预测工具,不仅快速准确,而且使研究人员在一次分析的基因数量上具有灵活性,而无需高性能计算集群。