Gómez-Pinedo Ulises, Sirerol-Piquer Maria Salomé, Durán-Moreno María, García-Verdugo José Manuel, Matias-Guiu Jorge
Neurobiology Laboratory, Neuroscience Institute, IdISSC, Hospital Clínico San Carlos, Universidad Complutense de Madrid, Madrid, Spain.
Laboratory of Comparative Neurobiology, Instituto Cavanilles de Biodiversidad y Biologia Evolutiva, Universidad de Valencia, Valencia, Spain.
Front Neurol. 2017 Jun 6;8:255. doi: 10.3389/fneur.2017.00255. eCollection 2017.
Alexander disease (AxD) is a rare disease caused by mutations in the gene encoding glial fibrillary acidic protein (GFAP). The disease is characterized by presence of GFAP aggregates in the cytoplasm of astrocytes and loss of myelin.
Determine the effect of AxD-related mutations on adult neurogenesis.
We transfected different types of mutant GFAP into neurospheres using the nucleofection technique.
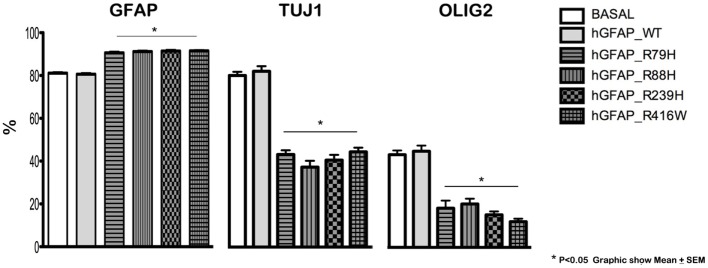
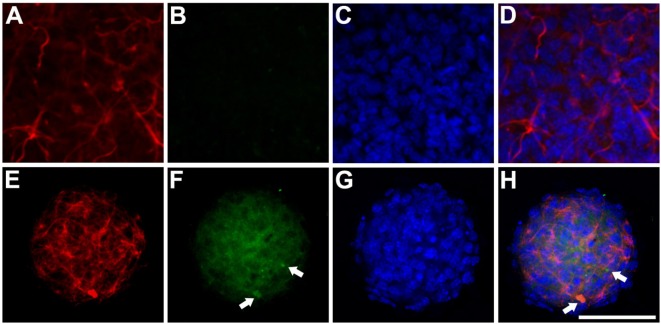
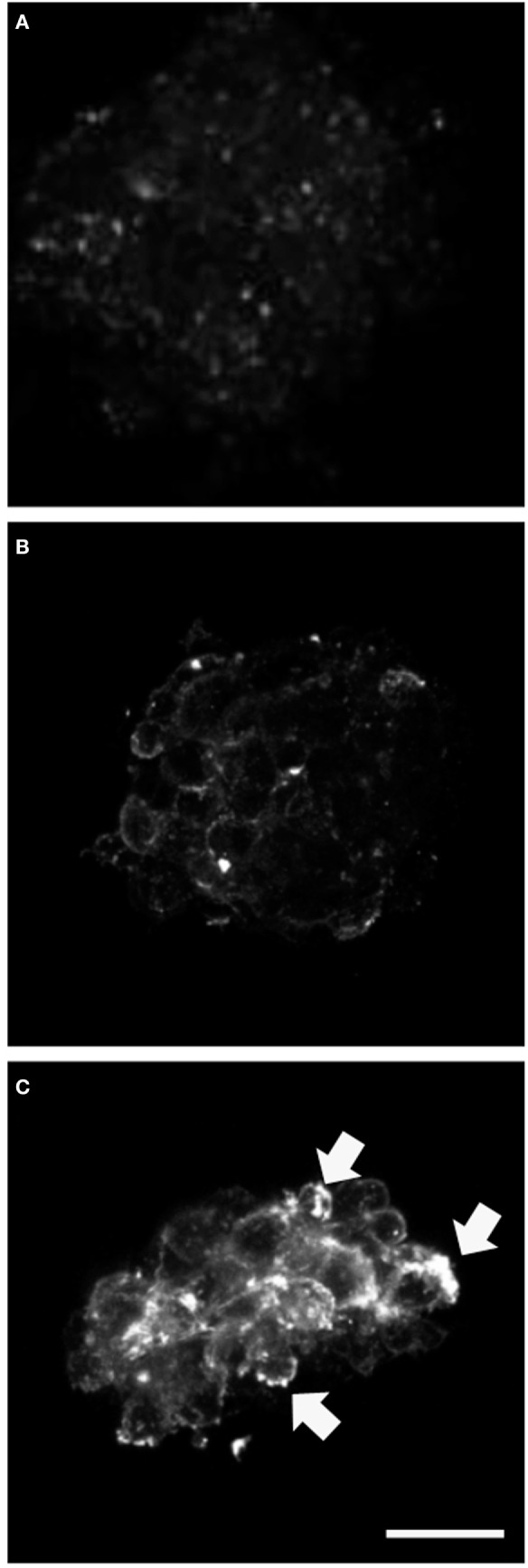
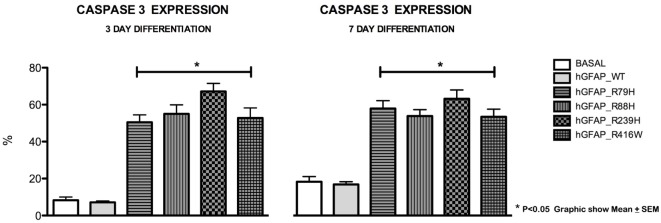
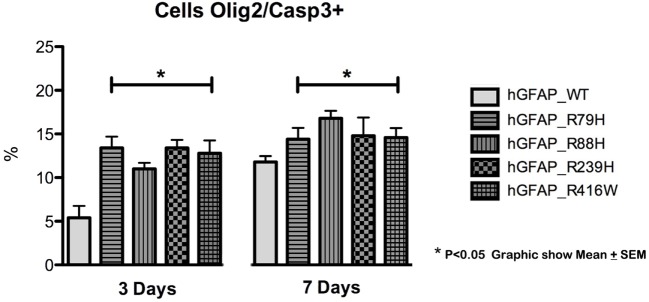
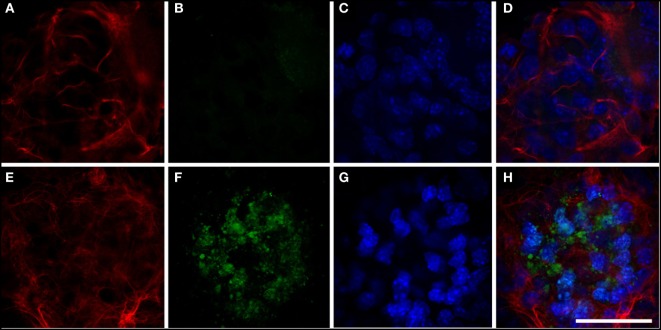
We find that mutations may cause coexpression of GFAP and NG2 in neurosphere cultures, which would inhibit the differentiation of precursors into oligodendrocytes and thus explain the myelin loss occurring in the disease. Transfection produces cells that differentiate into new cells marked simultaneously by GFAP and NG2 and whose percentage increased over days of differentiation. Increased expression of GFAP is due to a protein with an anomalous structure that forms aggregates throughout the cytoplasm of new cells. These cells display down-expression of vimentin and nestin. Up-expression of cathepsin D and caspase-3 in the first days of differentiation suggest that apoptosis as a lysosomal response may be at work. HSP27, a protein found in Rosenthal bodies, is expressed less at the beginning of the process although its presence increases in later stages.
Our findings seem to suggest that the mechanism of development of AxD may not be due to a function gain due to increase of GFAP, but to failure in the differentiation process may occur at the stage in which precursor cells transform into oligodendrocytes, and that possibility may provide the best explanation for the clinical and radiological images described in AxD.
亚历山大病(AxD)是一种由编码胶质纤维酸性蛋白(GFAP)的基因突变引起的罕见疾病。该疾病的特征是星形胶质细胞胞质中存在GFAP聚集体以及髓鞘缺失。
确定AxD相关突变对成体神经发生的影响。
我们使用核转染技术将不同类型的突变型GFAP转染到神经球中。
我们发现突变可能导致神经球培养物中GFAP和NG2共表达,这会抑制前体细胞向少突胶质细胞的分化,从而解释该疾病中发生的髓鞘缺失。转染产生的细胞分化为同时由GFAP和NG2标记的新细胞,其百分比在分化过程中随时间增加。GFAP表达增加是由于一种结构异常的蛋白质在新细胞的整个细胞质中形成聚集体。这些细胞显示波形蛋白和巢蛋白表达下调。分化初期组织蛋白酶D和半胱天冬酶-3的上调表明,作为溶酶体反应的细胞凋亡可能在起作用。热休克蛋白27(HSP27)是在罗森塔尔小体中发现的一种蛋白质,在该过程开始时表达较少,但其含量在后期增加。
我们的研究结果似乎表明,AxD的发病机制可能不是由于GFAP增加导致功能获得,而是在前体细胞转化为少突胶质细胞的阶段可能发生分化过程失败,这种可能性可能为AxD中描述的临床和放射学图像提供最佳解释。