Yang Xiaoyan, Shi Jing, Lei Haihong, Xia Bin, Mu Dezhi
Department of Pediatrics, West China Second University Hospital Key Laboratory of Obstetric & Gynaecologic and Pediatric Diseases and Birth Defects of Ministry of Education, Sichuan University, Chengdu, Sichuan, China.
Medicine (Baltimore). 2017 Jun;96(26):e7365. doi: 10.1097/MD.0000000000007365.
The carbamoyl phosphate synthetase I deficiency (CPS1D) was rare and hard to diagnose due to its atypical symptoms. Brain magnetic resonance imaging (MRI) was typically unavailable in other reports because most patients died before diagnosis was confirmed. Furthermore, it was found a new mutation that had not been described previously.
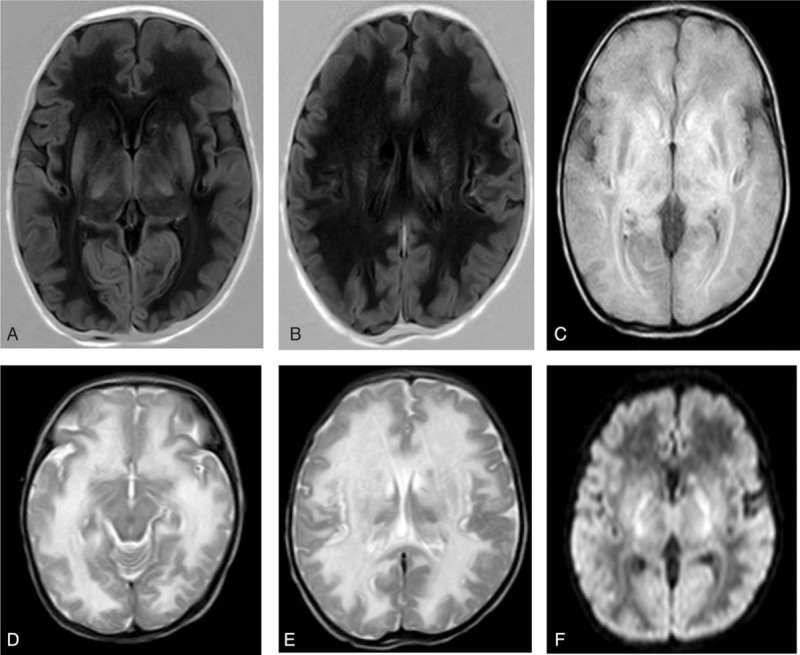
This is a case of neonatal-onset CPS1D with nonspecific clinical manifestations and deteriorating rapidly. Poor feeding, low activity, and tachypnoea were observed, with rapid progression on day 2 after birth. Severe systematic infection was considered first. However, blood culture and cerebrospinal fluid examination were negative. Symptoms were relief temporarily. Then seizure and tachypnoea reappeared as intravenous amino acids were provided. Further examination indicated severe hyperammonemia (serum ammonia level >500mmol/L). Brain MRI showed diffused white matter lesions.
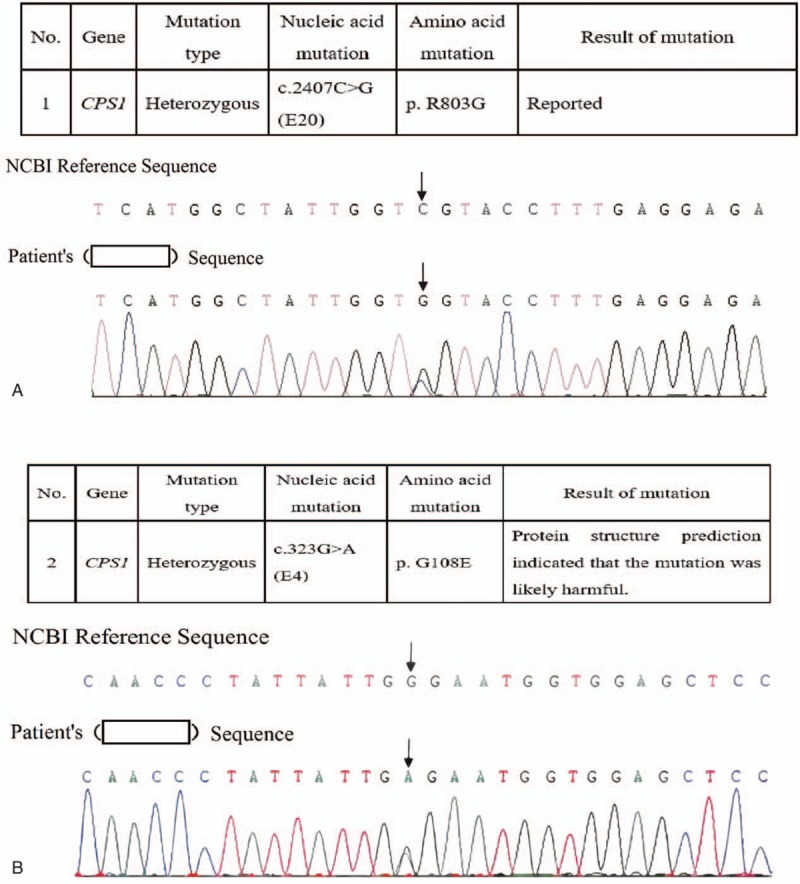
Genetic analysis revealed 2 heterozygous mutations in the CPS1 gene: c.2407C>G (p.803, R>G) in exon 20 and C.323G>A (p.108, G>E) in exon 4. The diagnosis of CSP1D was confirmed.
Fasting, the withdrawal of amino acids and plans to treat hyperammonemia were immediately implemented.
The parents decided to discontinue medical care.
Many CPS1D patients died before the diagnoses are confirmed due to its sudden onset, rapid deterioration, atypical symptoms, and low morbidity. Once hyperammonemia is confirmed, blood and urea amino acid analysis in combination with genetic examinations should be performed as early as possible, this approach would help establish diagnoses at an early stage and thus contribute to reducing mortality and improving prognosis.
氨甲酰磷酸合成酶I缺乏症(CPS1D)较为罕见,因其症状不典型而难以诊断。在其他报告中,脑部磁共振成像(MRI)通常无法进行,因为大多数患者在确诊前就已死亡。此外,还发现了一种先前未描述过的新突变。
这是一例新生儿期发病的CPS1D病例,临床表现不具特异性且病情迅速恶化。观察到喂养困难、活动减少和呼吸急促,出生后第2天病情迅速进展。首先考虑严重的系统性感染。然而,血培养和脑脊液检查均为阴性。症状暂时缓解。随后在提供静脉氨基酸时癫痫发作和呼吸急促再次出现。进一步检查显示严重高氨血症(血清氨水平>500mmol/L)。脑部MRI显示弥漫性白质病变。
基因分析显示CPS1基因存在2个杂合突变:外显子20中的c.2407C>G(p.803,R>G)和外显子4中的C.323G>A(p.108,G>E)。CSP1D诊断得以确诊。
立即实施禁食、停用氨基酸并制定治疗高氨血症的计划。
患儿父母决定停止医疗护理。
许多CPS1D患者因起病突然、病情迅速恶化、症状不典型及发病率低,在确诊前就已死亡。一旦确诊高氨血症,应尽早进行血液和尿素氨基酸分析并结合基因检查,这种方法有助于早期诊断,从而降低死亡率并改善预后。