Govaert Elisabeth, Van Steendam Katleen, Willems Sander, Vossaert Liesbeth, Dhaenens Maarten, Deforce Dieter
Laboratory of Pharmaceutical Biotechnology, Ghent University, Ghent, Belgium.
Proteomics. 2017 Aug;17(15-16). doi: 10.1002/pmic.201700052.
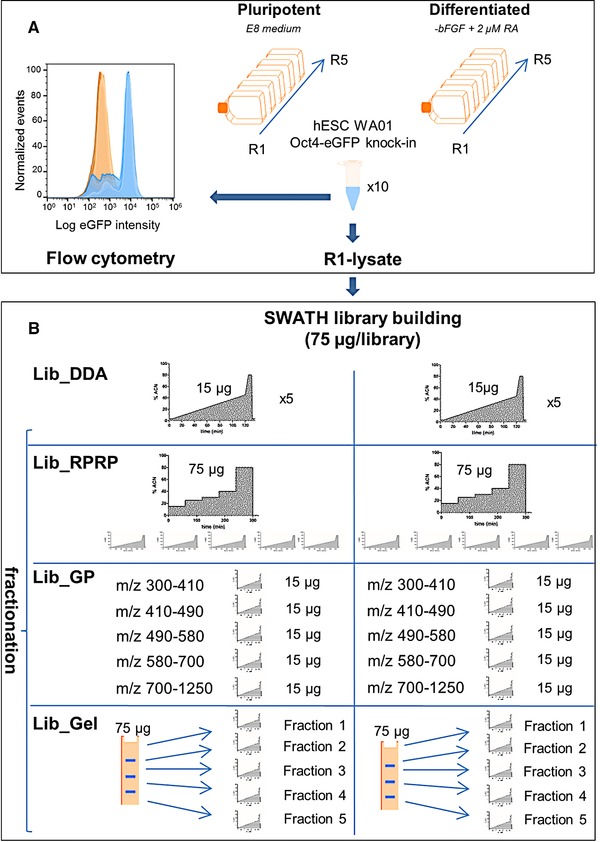
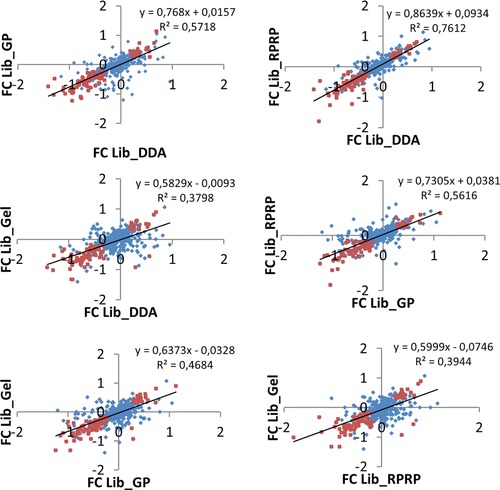
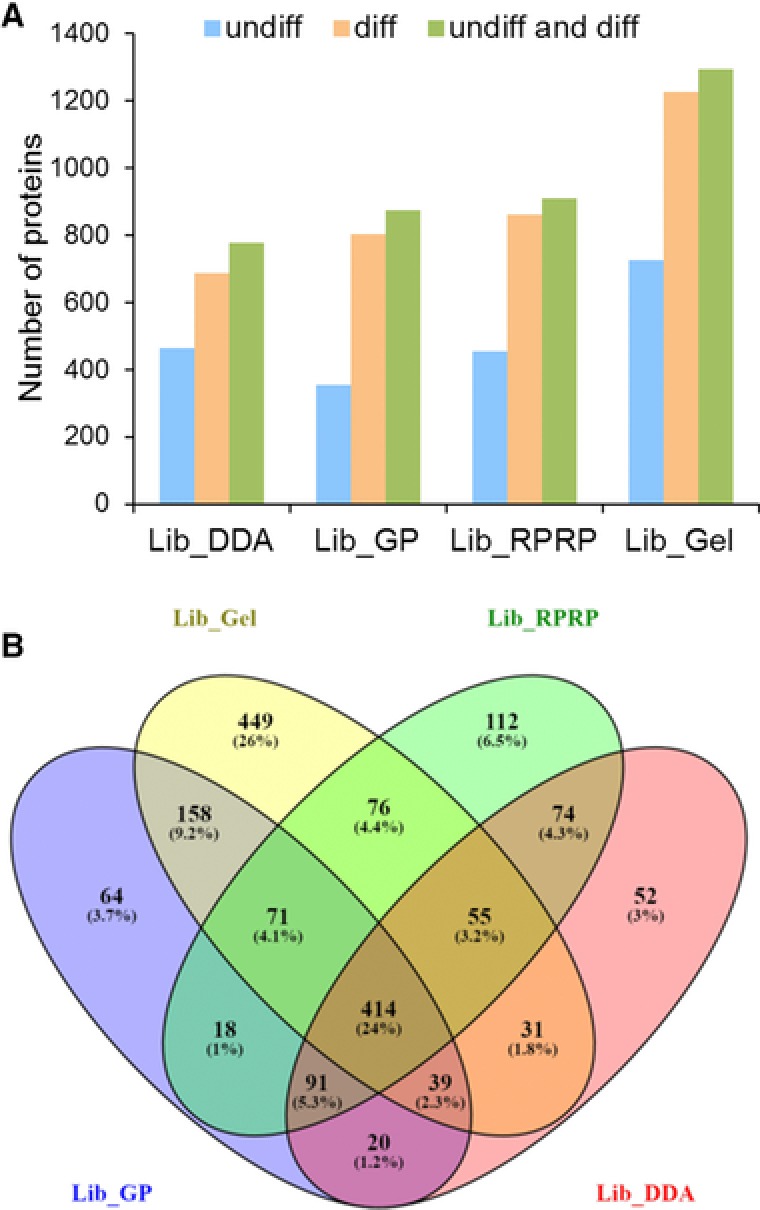
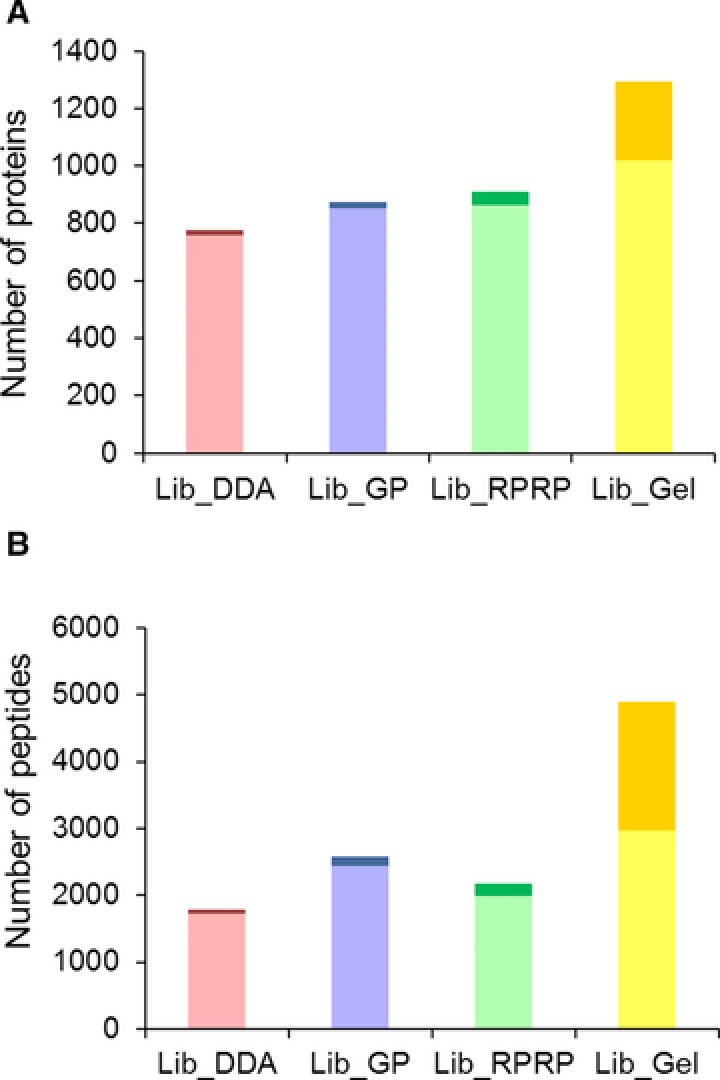
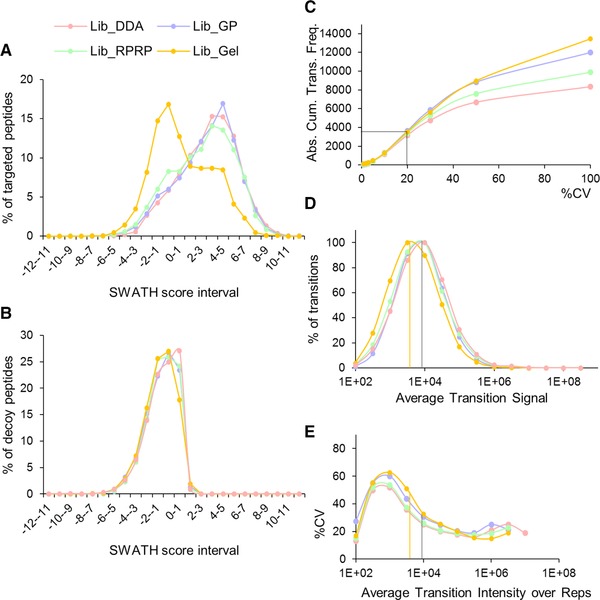
For data-independent acquisition by means of sequential window acquisition of all theoretical fragment ion spectra (SWATH), a reference library of data-dependent acquisition (DDA) runs is typically used to correlate the quantitative data from the fragment ion spectra with peptide identifications. The quality and coverage of such a reference library is therefore essential when processing SWATH data. In general, library sizes can be increased by reducing the impact of DDA precursor selection with replicate runs or fractionation. However, these strategies can affect the match between the library and SWATH measurement, and thus larger library sizes do not necessarily correspond to improved SWATH quantification. Here, three fractionation strategies to increase local library size were compared to standard library building using replicate DDA injection: protein SDS-PAGE fractionation, peptide high-pH RP-HPLC fractionation and MS-acquisition gas phase fractionation. The impact of these libraries on SWATH performance was evaluated in terms of the number of extracted peptides and proteins, the match quality of the peptides and the extraction reproducibility of the transitions. These analyses were conducted using the hydrophilic proteome of differentiating human embryonic stem cells. Our results show that SWATH quantitative results and interpretations are affected by choice of fractionation technique. Data are available via ProteomeXchange with identifier PXD006190.
对于通过所有理论碎片离子光谱的顺序窗口采集(SWATH)进行的数据非依赖型采集,通常使用数据依赖型采集(DDA)运行的参考库,以便将来自碎片离子光谱的定量数据与肽段鉴定相关联。因此,在处理SWATH数据时,这样一个参考库的质量和覆盖范围至关重要。一般来说,可以通过重复运行或分级分离来减少DDA前体选择的影响,从而增加库的大小。然而,这些策略可能会影响库与SWATH测量之间的匹配,因此更大的库大小不一定对应于改进的SWATH定量。在这里,将三种增加局部库大小的分级分离策略与使用重复DDA进样的标准库构建方法进行了比较:蛋白质SDS-PAGE分级分离、肽段高pH值反相高效液相色谱(RP-HPLC)分级分离和质谱采集气相分级分离。根据提取的肽段和蛋白质数量、肽段的匹配质量以及跃迁的提取重现性,评估了这些库对SWATH性能的影响。这些分析是使用分化中的人类胚胎干细胞的亲水性蛋白质组进行的。我们的结果表明,SWATH定量结果和解释受分级分离技术选择的影响。数据可通过ProteomeXchange获得,标识符为PXD006190。