Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore, Block MD9, 2 Medical Drive, Singapore, 117 597, Singapore.
Neurobiology/Aging Program, Life Sciences Institute (LSI), National University of Singapore, #04-44, 28 Medical Drive, Singapore, 117 456, Singapore.
Aging Cell. 2017 Oct;16(5):1062-1072. doi: 10.1111/acel.12634. Epub 2017 Jun 30.
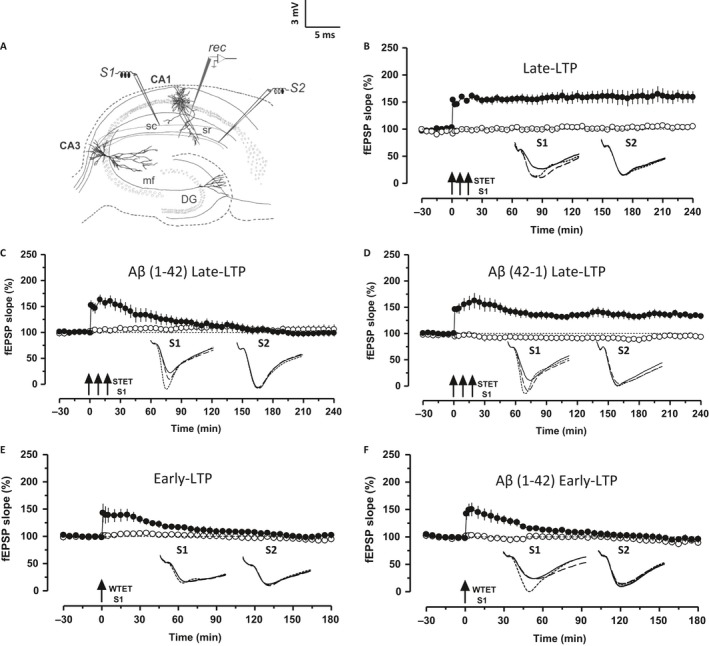
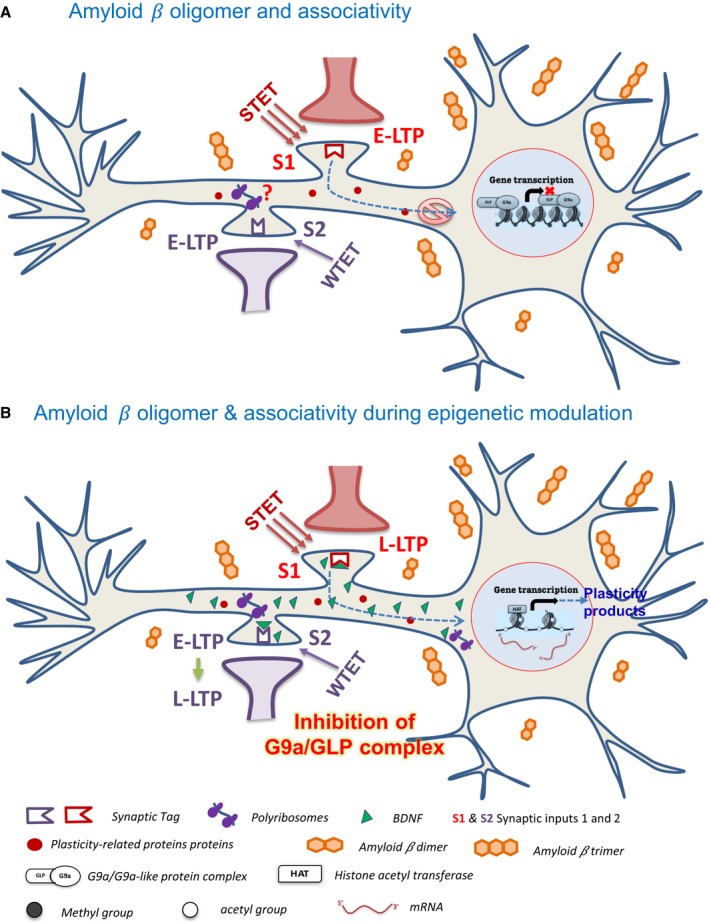
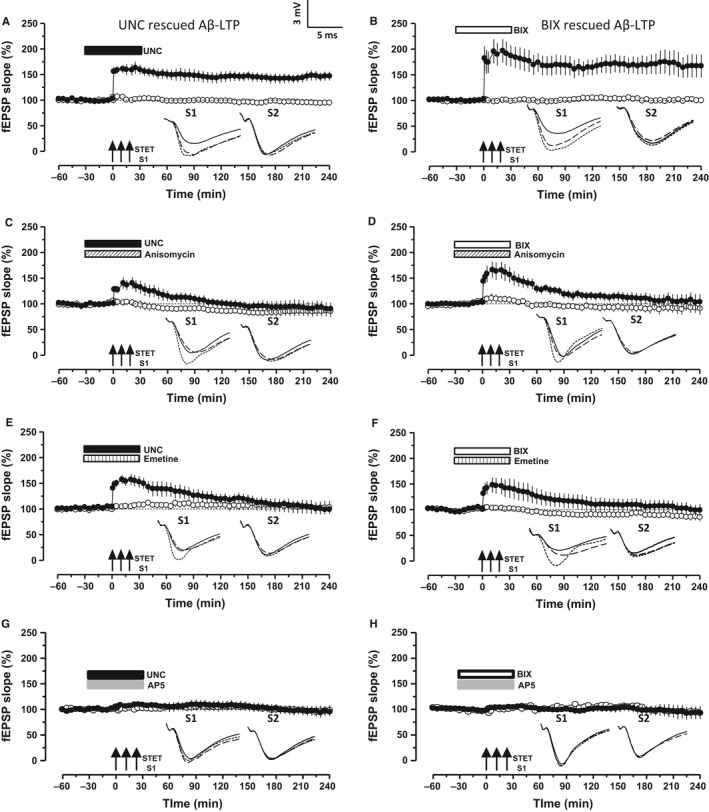
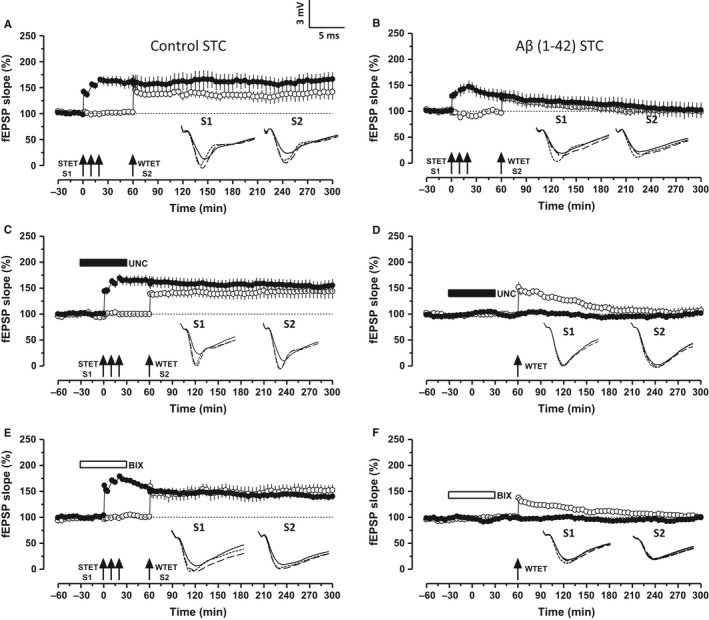
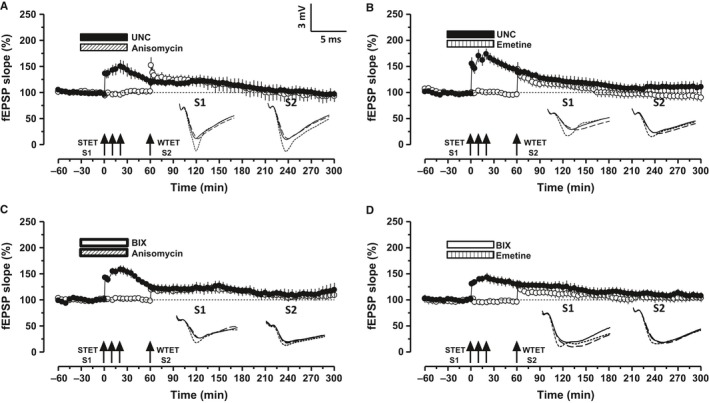
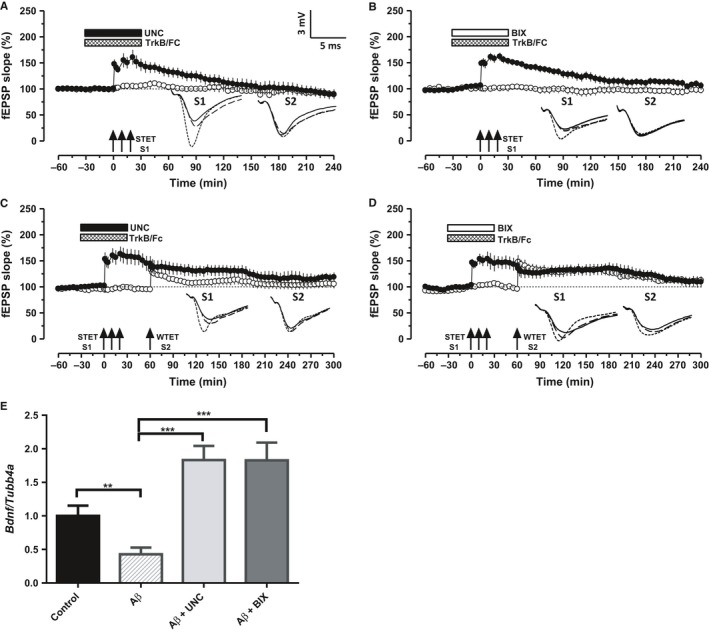
Altered epigenetic mechanisms are implicated in the cognitive decline associated with neurodegenerative diseases such as in Alzheimer's disease (AD). AD is the most prevalent form of dementia worldwide; amyloid plaques and neurofibrillary tangles are the histopathological hallmarks of AD. We have recently reported that the inhibition of G9a/GLP complex promotes long-term potentiation (LTP) and its associative mechanisms such as synaptic tagging and capture (STC). However, the role of this complex in plasticity impairments remains elusive. Here, we investigated the involvement of G9a/GLP complex in alleviating the effects of soluble Amyloid-β 1-42 oligomers (oAβ) on neuronal plasticity and associativity in the CA1 region of acute hippocampal slices from 5- to 7-week-old male Wistar rats. Our findings demonstrate that the regulation of G9a/GLP complex by inhibiting its catalytic activity reverses the amyloid-β oligomer-induced deficits in late-LTP and STC. This is achieved by releasing the transcription repression of the brain-derived neurotrophic factor (Bdnf) gene. The catalytic inhibition of G9a/GLP complex leads to the upregulation of Bdnf expression in the slices treated with oAβ. This further ensures the availability of BDNF that subsequently binds its receptor tyrosine kinase B (TrkB) and maintains the late-LTP. Furthermore, the capture of BDNF by weakly activated synapses re-establishes STC. Our findings regarding the reinstatement of functional plasticity and associativity in AD-like conditions provide the first evidence for the role of G9a/GLP complex in AD. We propose G9a/GLP complex as the possible target for preventing oAβ-induced plasticity deficits in hippocampal neurons.
表观遗传机制的改变与神经退行性疾病(如阿尔茨海默病)相关的认知能力下降有关。AD 是全球最常见的痴呆形式;淀粉样斑块和神经原纤维缠结是 AD 的组织病理学特征。我们最近报道,G9a/GLP 复合物的抑制可促进长时程增强(LTP)及其相关机制,如突触标记和捕获(STC)。然而,该复合物在可塑性损伤中的作用仍不清楚。在这里,我们研究了 G9a/GLP 复合物在缓解可溶性淀粉样β 1-42 寡聚体(oAβ)对急性海马切片 CA1 区神经元可塑性和关联性的影响中的作用。我们的研究结果表明,通过抑制其催化活性调节 G9a/GLP 复合物可逆转 oAβ 诱导的晚期长时程增强(LTP)和 STC 缺陷。这是通过释放脑源性神经营养因子(BDNF)基因的转录抑制来实现的。G9a/GLP 复合物的催化抑制导致 oAβ 处理的切片中 Bdnf 表达上调。这进一步确保了 BDNF 的可用性,随后 BDNF 与其受体酪氨酸激酶 B(TrkB)结合并维持晚期 LTP。此外,BDNF 通过弱激活的突触被捕获,从而重新建立 STC。我们在 AD 样条件下恢复功能性可塑性和关联性的发现为 G9a/GLP 复合物在 AD 中的作用提供了第一个证据。我们提出 G9a/GLP 复合物是预防 oAβ 诱导的海马神经元可塑性缺陷的可能靶点。