Weissman Daniel B, Hallatschek Oskar
Department of Physics, Emory University, Atlanta, United States.
Department of Physics and Integrative Biology, University of California, Berkeley, Berkeley, United States.
Elife. 2017 Jul 3;6:e24836. doi: 10.7554/eLife.24836.
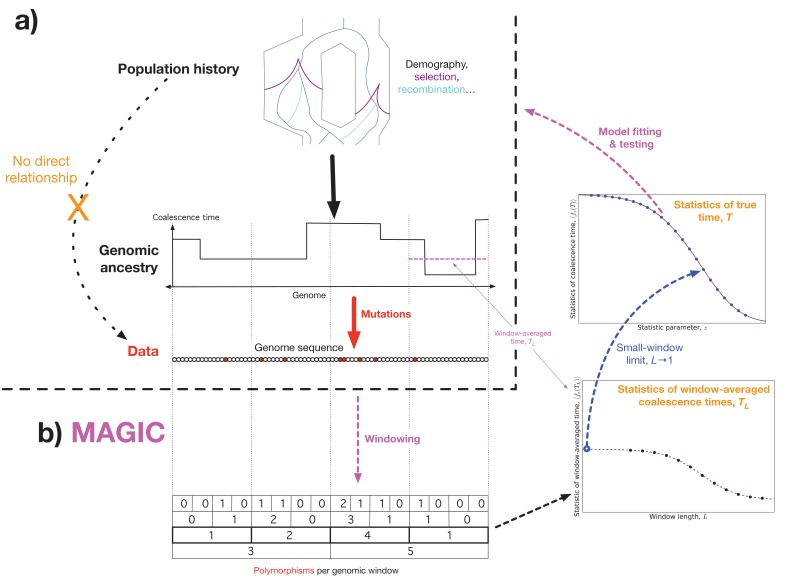
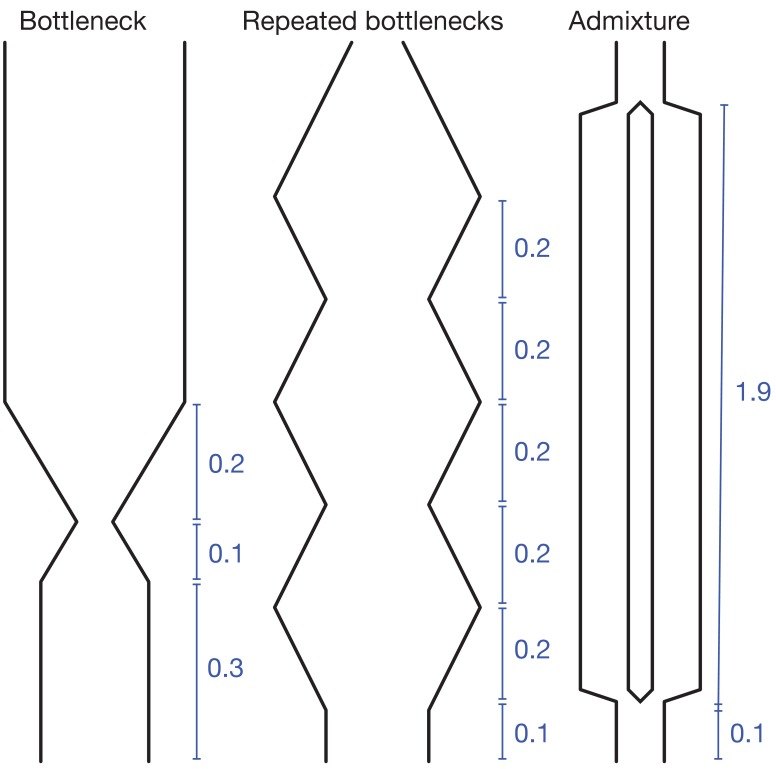
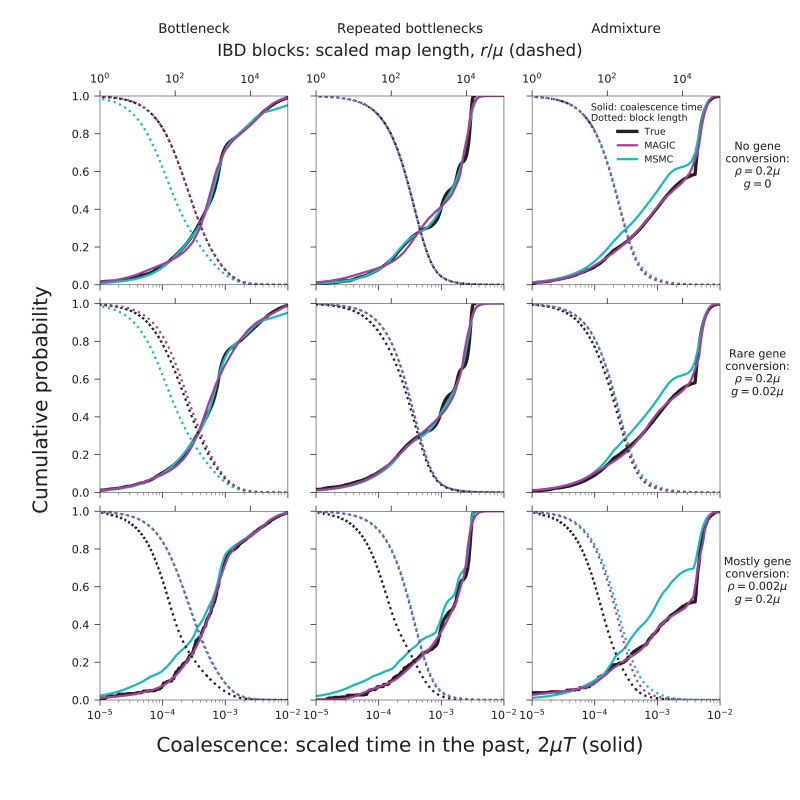
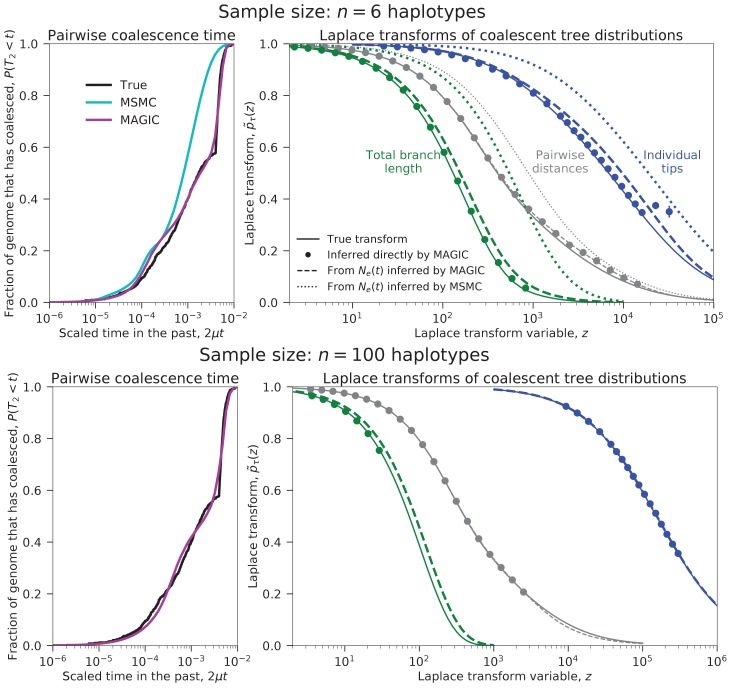
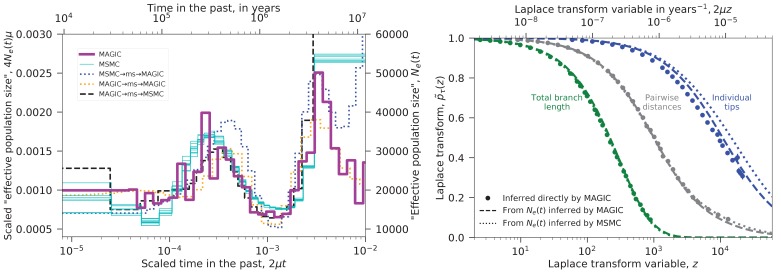
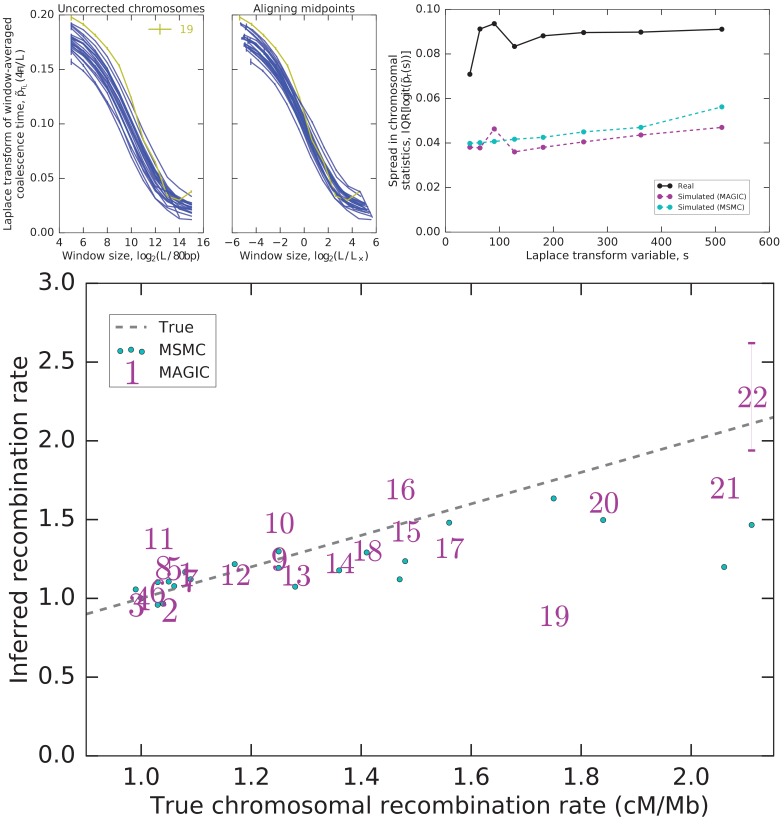
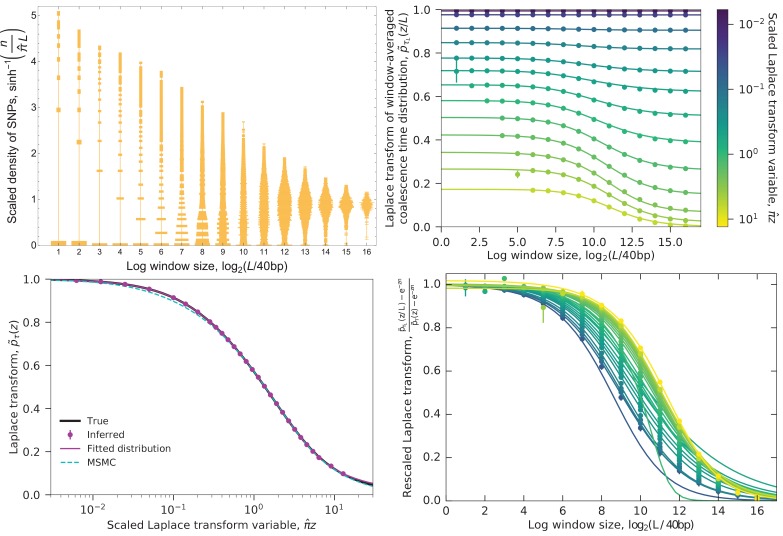
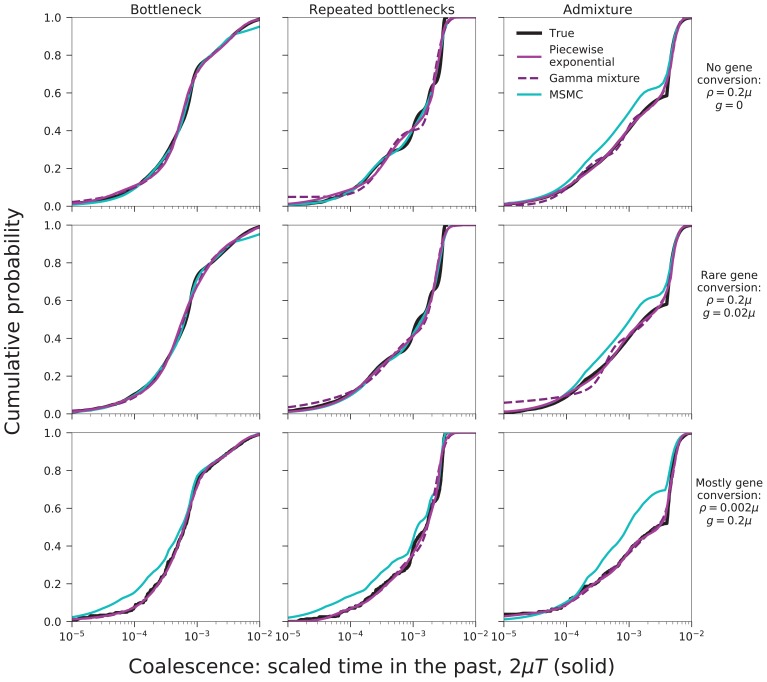
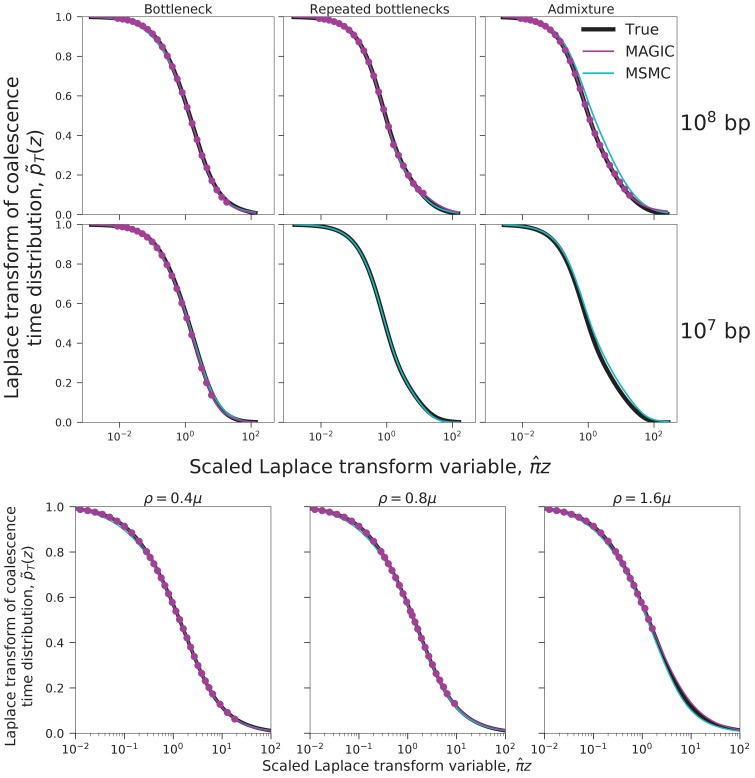
Samples of multiple complete genome sequences contain vast amounts of information about the evolutionary history of populations, much of it in the associations among polymorphisms at different loci. We introduce a method, Minimal-Assumption Genomic Inference of Coalescence (MAGIC), that reconstructs key features of the evolutionary history, including the distribution of coalescence times, by integrating information across genomic length scales without using an explicit model of coalescence or recombination, allowing it to analyze arbitrarily large samples without phasing while making no assumptions about ancestral structure, linked selection, or gene conversion. Using simulated data, we show that the performance of MAGIC is comparable to that of PSMC' even on single diploid samples generated with standard coalescent and recombination models. Applying MAGIC to a sample of human genomes reveals evidence of non-demographic factors driving coalescence.
多个完整基因组序列的样本包含了大量关于种群进化历史的信息,其中大部分信息存在于不同位点多态性之间的关联中。我们引入了一种方法,即最小假设基因组合并推断法(MAGIC),该方法通过整合基因组不同长度尺度上的信息来重建进化历史的关键特征,包括合并时间的分布,而无需使用明确的合并或重组模型,从而能够在不进行定相的情况下分析任意大的样本,同时不对祖先结构、连锁选择或基因转换做出任何假设。使用模拟数据,我们表明即使在使用标准合并和重组模型生成的单倍体样本上,MAGIC的性能也与PSMC相当。将MAGIC应用于人类基因组样本揭示了驱动合并的非人口统计学因素的证据。