Rocklin Gabriel J, Chidyausiku Tamuka M, Goreshnik Inna, Ford Alex, Houliston Scott, Lemak Alexander, Carter Lauren, Ravichandran Rashmi, Mulligan Vikram K, Chevalier Aaron, Arrowsmith Cheryl H, Baker David
Department of Biochemistry and Institute for Protein Design, University of Washington, Seattle, WA 98195, USA.
Graduate Program in Biological Physics, Structure, and Design, University of Washington, Seattle, WA 98195, USA.
Science. 2017 Jul 14;357(6347):168-175. doi: 10.1126/science.aan0693.
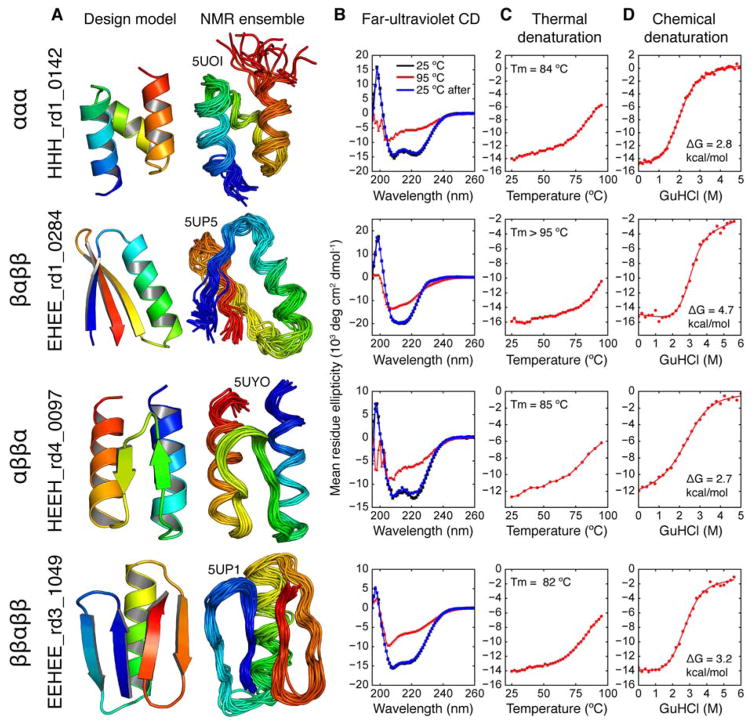
Proteins fold into unique native structures stabilized by thousands of weak interactions that collectively overcome the entropic cost of folding. Although these forces are "encoded" in the thousands of known protein structures, "decoding" them is challenging because of the complexity of natural proteins that have evolved for function, not stability. We combined computational protein design, next-generation gene synthesis, and a high-throughput protease susceptibility assay to measure folding and stability for more than 15,000 de novo designed miniproteins, 1000 natural proteins, 10,000 point mutants, and 30,000 negative control sequences. This analysis identified more than 2500 stable designed proteins in four basic folds-a number sufficient to enable us to systematically examine how sequence determines folding and stability in uncharted protein space. Iteration between design and experiment increased the design success rate from 6% to 47%, produced stable proteins unlike those found in nature for topologies where design was initially unsuccessful, and revealed subtle contributions to stability as designs became increasingly optimized. Our approach achieves the long-standing goal of a tight feedback cycle between computation and experiment and has the potential to transform computational protein design into a data-driven science.
蛋白质折叠成独特的天然结构,由数千种弱相互作用稳定,这些相互作用共同克服了折叠的熵成本。尽管这些作用力“编码”在数千种已知的蛋白质结构中,但由于天然蛋白质是为功能而非稳定性进化而来,其复杂性使得“解码”这些作用力具有挑战性。我们结合了计算蛋白质设计、新一代基因合成和高通量蛋白酶敏感性测定,以测量超过15000种从头设计的微型蛋白质、1000种天然蛋白质、10000个点突变体和30000个阴性对照序列的折叠和稳定性。该分析在四种基本折叠中鉴定出2500多种稳定的设计蛋白质——这一数量足以使我们能够系统地研究序列如何在未知的蛋白质空间中决定折叠和稳定性。设计与实验之间的迭代将设计成功率从6%提高到47%,产生了与自然界中不同的稳定蛋白质,用于最初设计不成功的拓扑结构,并随着设计的日益优化揭示了对稳定性的微妙贡献。我们的方法实现了计算与实验之间紧密反馈循环的长期目标,并有潜力将计算蛋白质设计转变为一门数据驱动的科学。