Department of Pharmacology, Northwestern University Feinberg School of Medicine, Chicago, IL, USA.
Center for Synthetic Biology, Northwestern University, Evanston, IL, USA.
Nature. 2023 Aug;620(7973):434-444. doi: 10.1038/s41586-023-06328-6. Epub 2023 Jul 19.
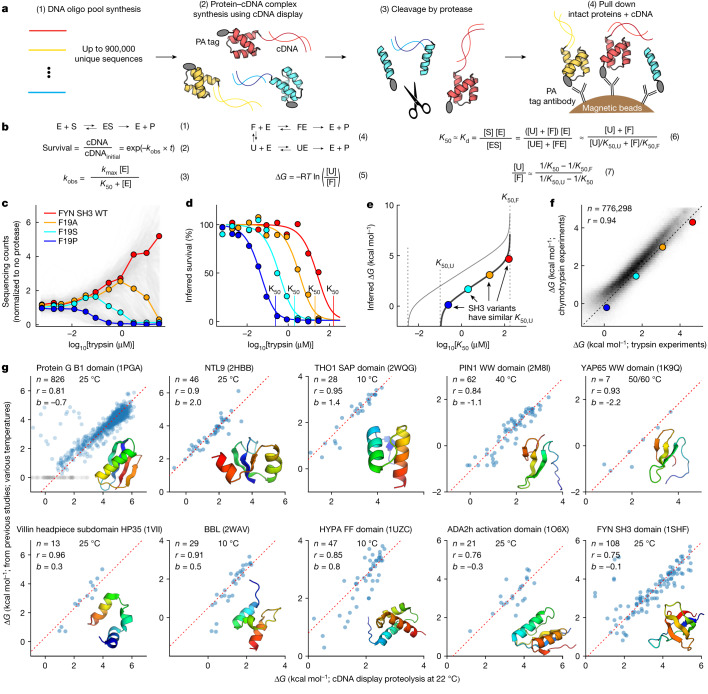
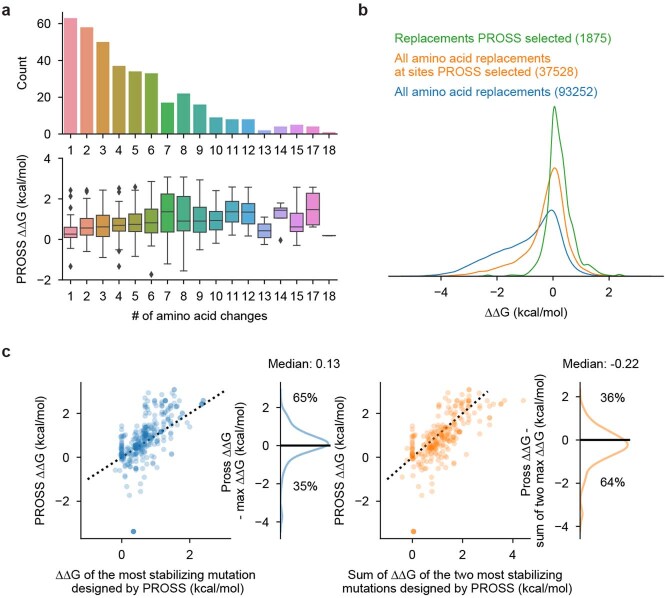
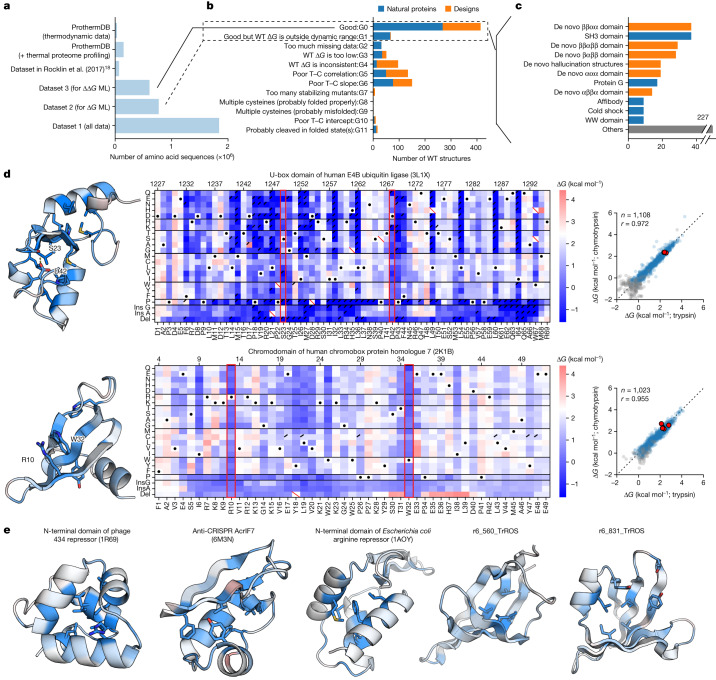
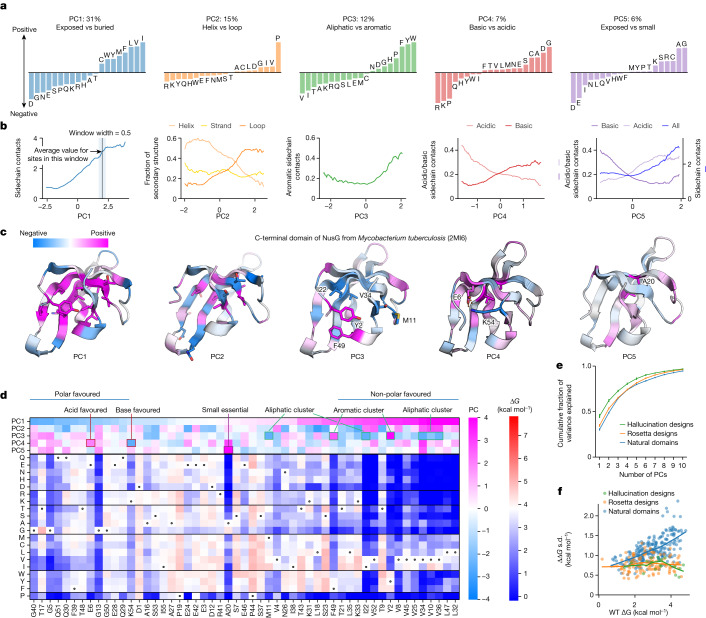
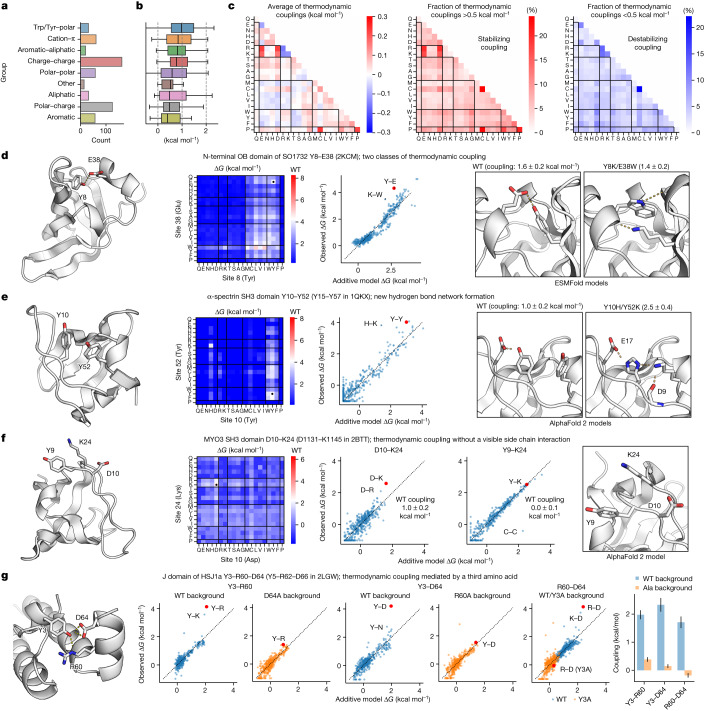
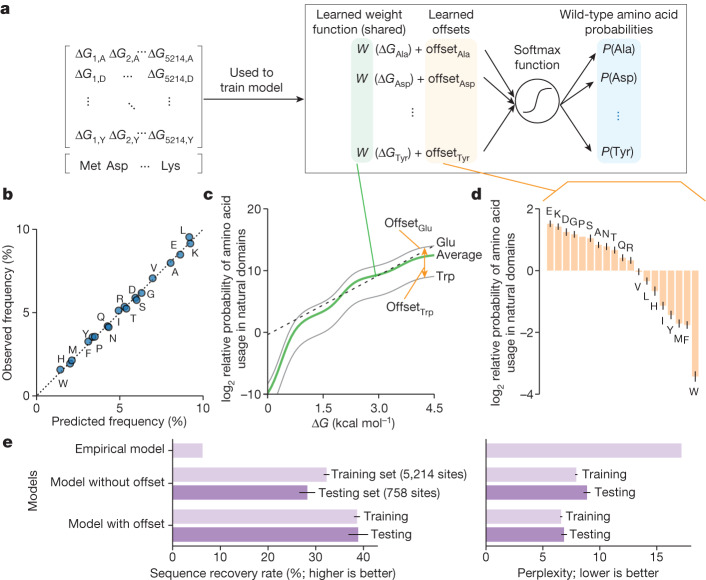
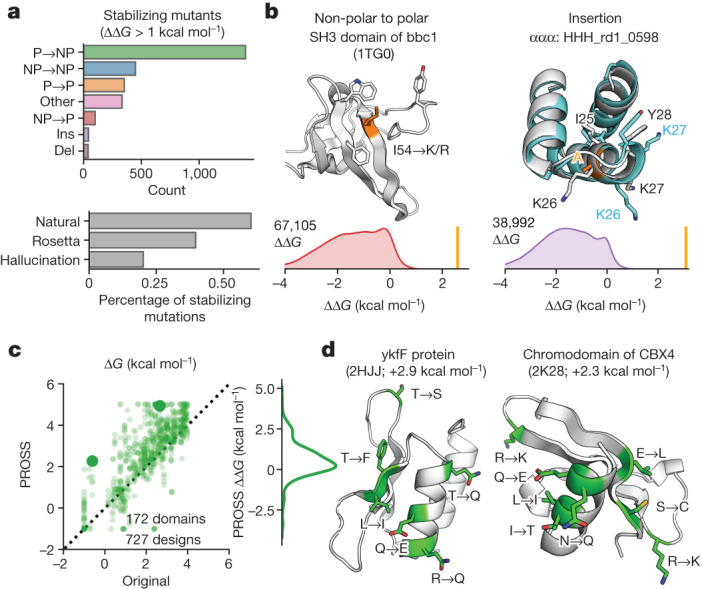
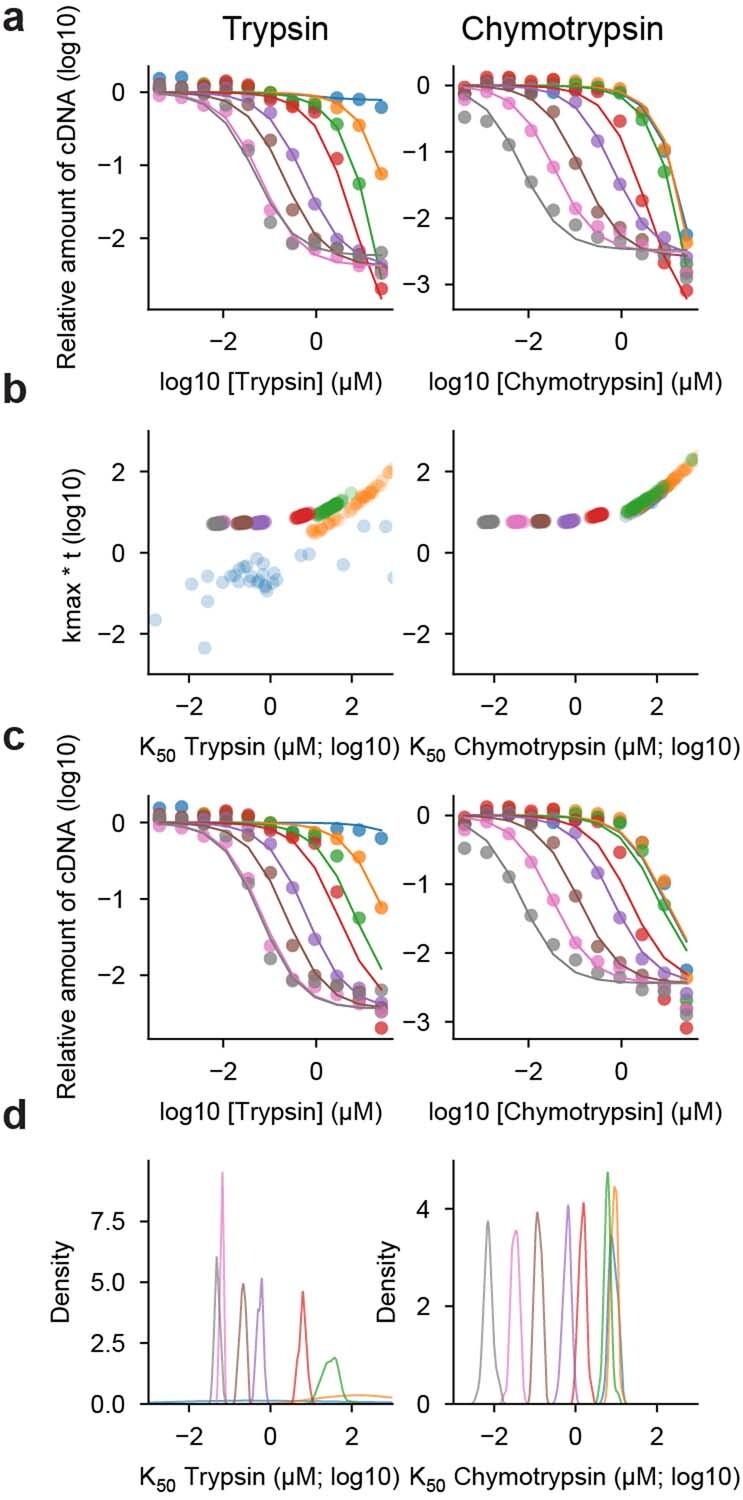
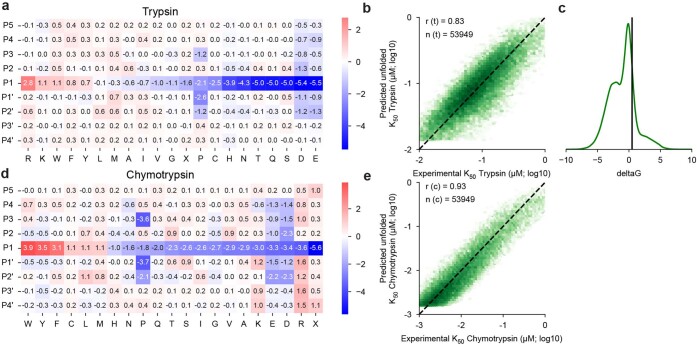
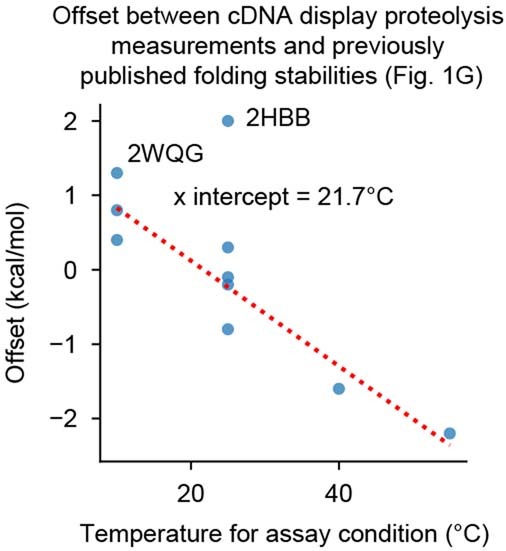
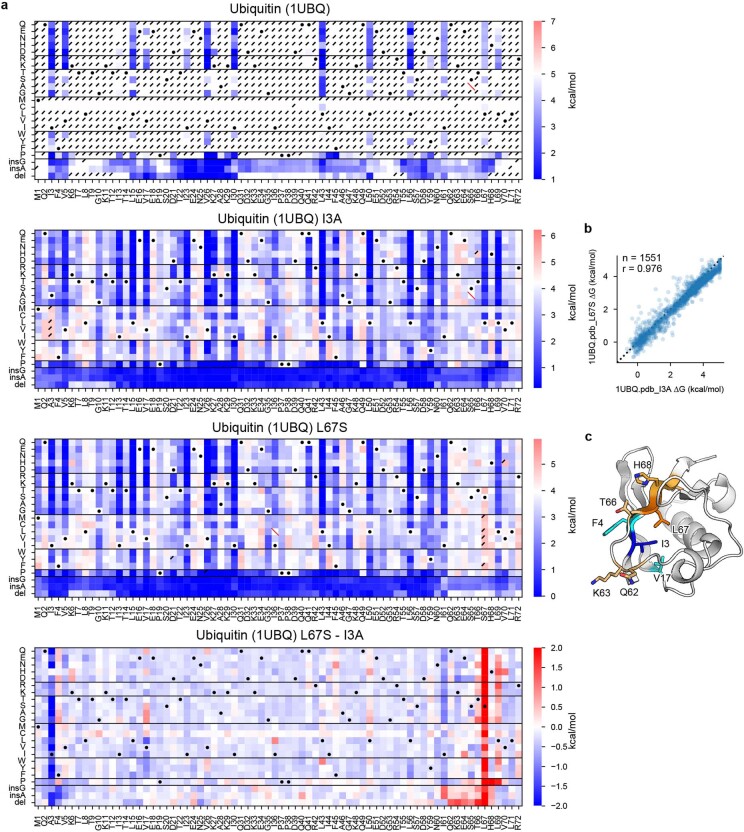
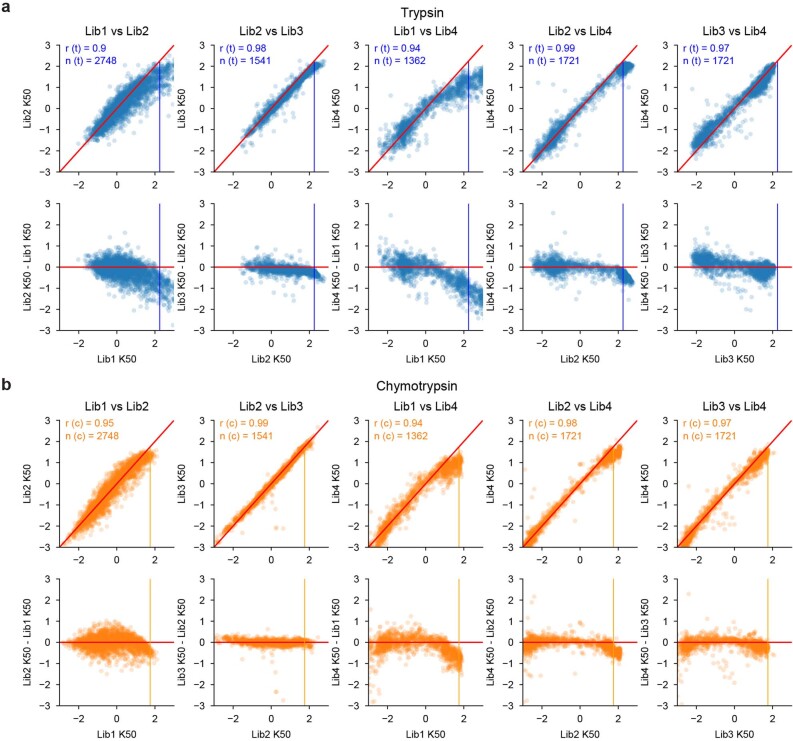
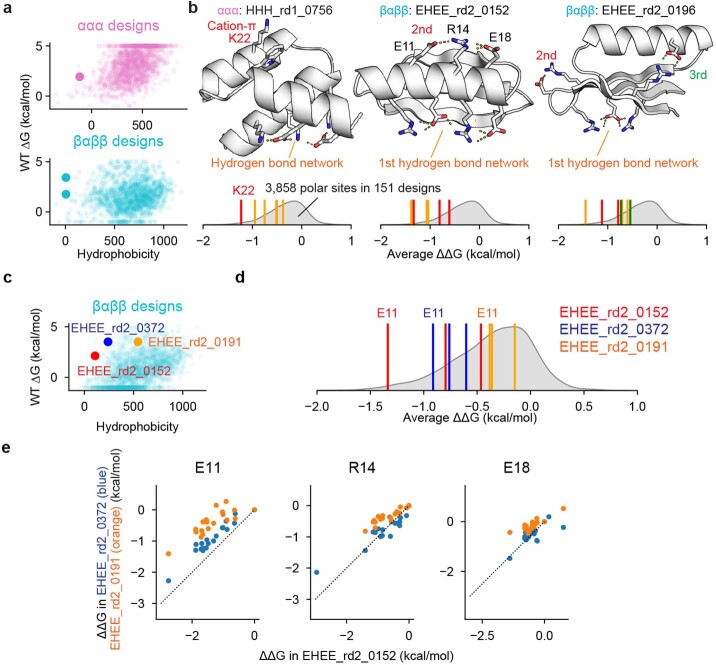
Advances in DNA sequencing and machine learning are providing insights into protein sequences and structures on an enormous scale. However, the energetics driving folding are invisible in these structures and remain largely unknown. The hidden thermodynamics of folding can drive disease, shape protein evolution and guide protein engineering, and new approaches are needed to reveal these thermodynamics for every sequence and structure. Here we present cDNA display proteolysis, a method for measuring thermodynamic folding stability for up to 900,000 protein domains in a one-week experiment. From 1.8 million measurements in total, we curated a set of around 776,000 high-quality folding stabilities covering all single amino acid variants and selected double mutants of 331 natural and 148 de novo designed protein domains 40-72 amino acids in length. Using this extensive dataset, we quantified (1) environmental factors influencing amino acid fitness, (2) thermodynamic couplings (including unexpected interactions) between protein sites, and (3) the global divergence between evolutionary amino acid usage and protein folding stability. We also examined how our approach could identify stability determinants in designed proteins and evaluate design methods. The cDNA display proteolysis method is fast, accurate and uniquely scalable, and promises to reveal the quantitative rules for how amino acid sequences encode folding stability.
DNA 测序和机器学习的进步正在大规模地提供对蛋白质序列和结构的深入了解。然而,这些结构中无法看到驱动折叠的能量学,并且这些能量学在很大程度上仍然未知。折叠的隐藏热力学可以引发疾病、塑造蛋白质进化并指导蛋白质工程,因此需要新的方法来揭示每个序列和结构的这些热力学。在这里,我们提出了 cDNA 显示蛋白水解,这是一种在一周的实验中测量多达 90 万个蛋白质结构域热力学折叠稳定性的方法。通过总共 180 万次测量,我们整理了大约 776,000 个高质量的折叠稳定性数据集,涵盖了所有单个氨基酸变体和 331 个天然和 148 个从头设计的蛋白质结构域的 40-72 个氨基酸的选择双突变体。使用这个广泛的数据集,我们量化了:(1) 影响氨基酸适应性的环境因素,(2) 蛋白质位点之间的热力学耦合(包括意外的相互作用),以及 (3) 进化氨基酸使用和蛋白质折叠稳定性之间的全球差异。我们还研究了我们的方法如何识别设计蛋白质中的稳定性决定因素并评估设计方法。cDNA 显示蛋白水解方法快速、准确且具有独特的可扩展性,有望揭示氨基酸序列如何编码折叠稳定性的定量规则。