Chen Xingchun, Wang Lijun, Zhou Jiali, Wu Honglong, Li Dong, Cui Yanchao, Lu Binghuai
Department of Laboratory Medicine, People's Hospital of Guangxi Zhuang Autonomous Region, Nanning, 530021, China.
Department of Laboratory Medicine, Beijing Tsinghua Chang Gung Hospital, Tsinghua University, Beijing, 102218, China.
BMC Infect Dis. 2017 Jul 21;17(1):508. doi: 10.1186/s12879-017-2616-1.
Bacterial species belonging to the genus Exiguobacterium are facultative anaerobic, non-spore-forming, Gram-positive bacilli, and rarely associated with human infections. Herein, we reported the first case of community-acquired pneumonia (CAP) and bacteremia due to Exiguobacterium spp. in China.
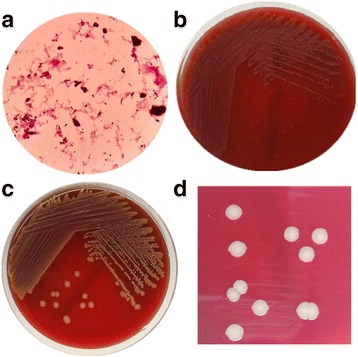
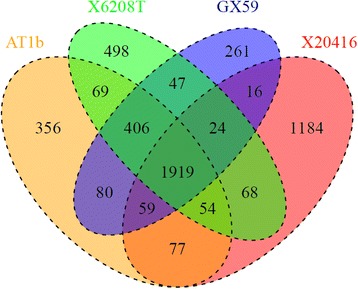
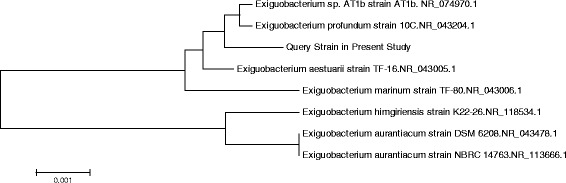
An adult male with severe CAP was hospitalized. The pathogen was isolated from his bloodstream and broncho-alveolar lavage fluid. The correct identification of the micro-organism was achieved using 16S rRNA sequencing, and its antibiotic susceptibility test was performed by microdilution method. The Whole Genome Sequencing (WGS) was used to characterize its genetic features and to elucidate its potential pathogenic mechanisms. Furthermore, its genome sequence was also compared with those of 3 publicly-available Exiguobacterium strains. A PubMed search was performed for further understanding the features of Exiguobacterium infections. Phylogenetic analysis of the 16S rRNA gene sequence showed that the strain GX59 was most closely related to Exiguobacterium AT1b (99.7%). The genome of GX59 was 2,727,929 bp in size, harbouring 2855 putative protein-coding genes, 5 rRNA operons, 37 tRNA genes and 1 tmRNA. The multiple genome comparison of 4 Exiguobacterium strains demonstrated that Exiguobacterium contained 37 genes of secretion systems, including sec, tat, FEA, Type IV Pili and competence-related DNA transformation transporter (Com). Virulence factors of the micro-organism included tlyC, NprR, MCP, Dam, which might play a critical role in causing lethal infection.
The study highlighted the potential pathogenicity of the genus Exiguobacterium for its unique genes encoding various virulence factors and those associated with antibiotic resistance, therefore, its clinical significance should be valued.
嗜冷栖热放线菌属的细菌为兼性厌氧、无芽孢形成的革兰氏阳性杆菌,很少与人类感染相关。在此,我们报告了中国首例由嗜冷栖热放线菌属引起的社区获得性肺炎(CAP)和菌血症病例。
一名患有严重CAP的成年男性住院治疗。病原体从其血液和支气管肺泡灌洗液中分离出来。通过16S rRNA测序实现了微生物的正确鉴定,并通过微量稀释法进行了抗生素敏感性试验。使用全基因组测序(WGS)来表征其遗传特征并阐明其潜在的致病机制。此外,还将其基因组序列与3株公开可得的嗜冷栖热放线菌菌株的序列进行了比较。通过PubMed搜索以进一步了解嗜冷栖热放线菌感染的特征。16S rRNA基因序列的系统发育分析表明,菌株GX59与嗜冷栖热放线菌AT1b关系最为密切(99.7%)。GX59的基因组大小为2,727,929 bp,包含2855个推定的蛋白质编码基因、5个rRNA操纵子、37个tRNA基因和1个tmRNA。4株嗜冷栖热放线菌菌株的多基因组比较表明,嗜冷栖热放线菌含有37个分泌系统基因,包括sec、tat、FEA、IV型菌毛和与感受态相关的DNA转化转运体(Com)。该微生物的毒力因子包括tlyC、NprR、MCP、Dam,它们可能在导致致命感染中起关键作用。
该研究强调了嗜冷栖热放线菌属因其编码各种毒力因子和与抗生素耐药性相关的独特基因而具有潜在致病性,因此,应重视其临床意义。