Laboratory of Biochemistry and Vascular Biology, Center for Biologics Evaluation and Research, Food and Drug Administration, Silver Spring, MD 20993, USA.
Metallomics. 2017 Sep 20;9(9):1260-1270. doi: 10.1039/c7mt00104e.
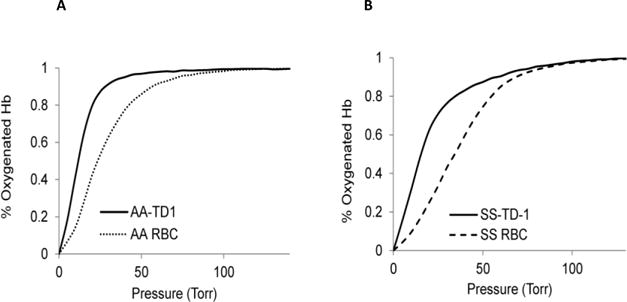
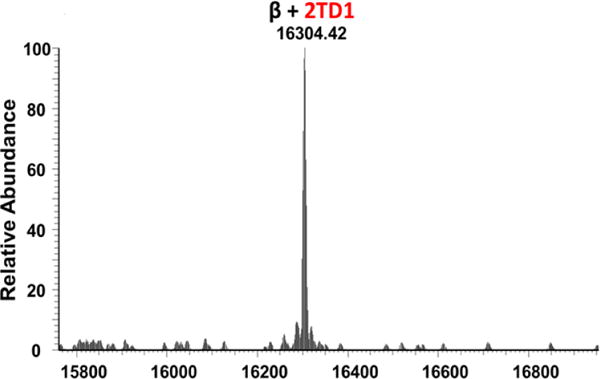
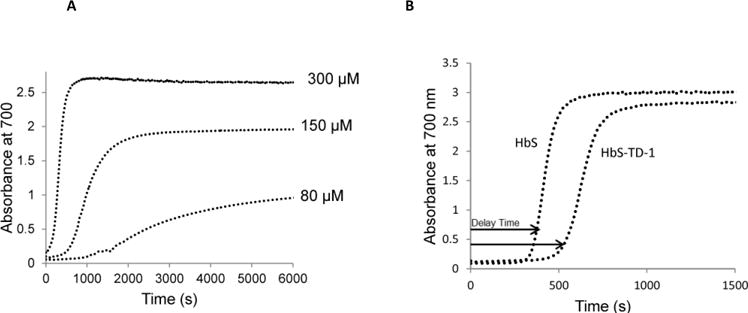
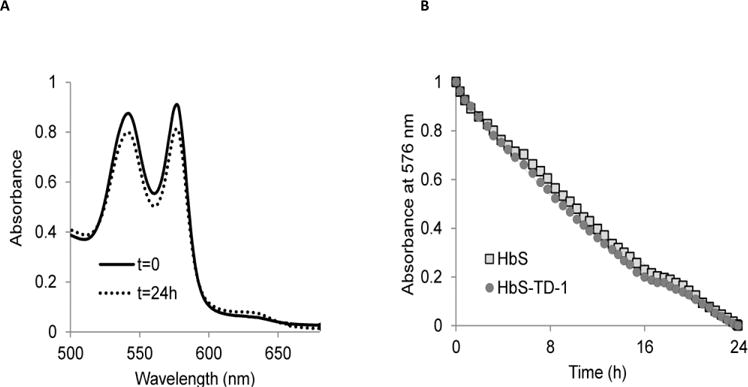
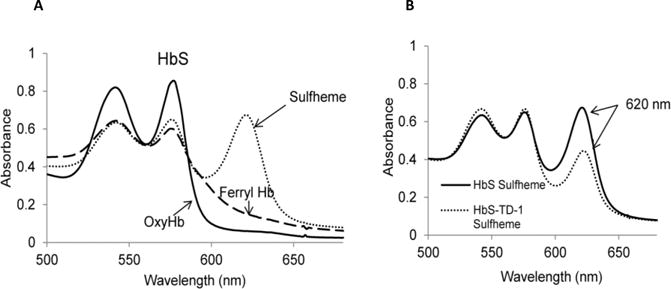
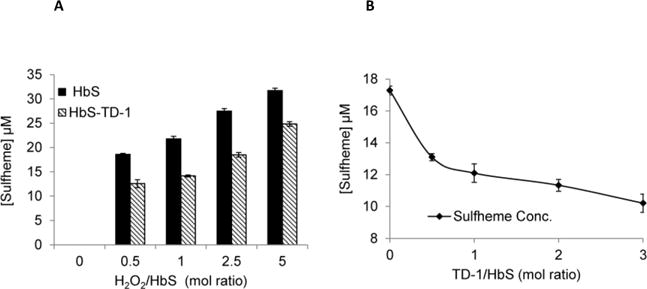
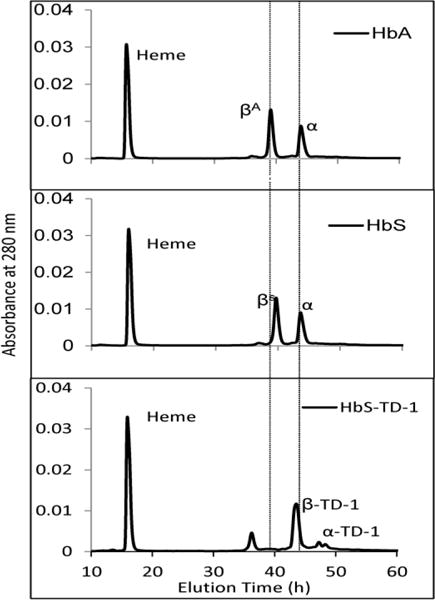
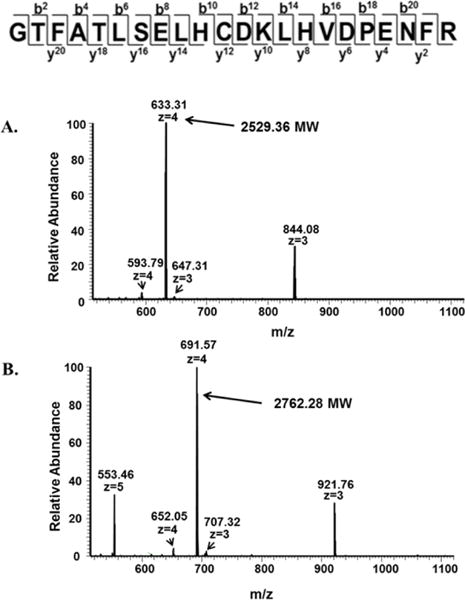
Sickle cell disease (SCD) is an inherited blood disorder caused by a β globin gene mutation of hemoglobin (HbS). The polymerization of deoxyHbS and its subsequent aggregation (into long fibers) is the primary molecular event which leads to red blood cell (RBC) sickling and ultimately hemolytic anemia. We have recently suggested that HbS oxidative toxicity may also contribute to SCD pathophysiology due to its defective pseudoperoxidase activity. As a consequence, a persistently higher oxidized ferryl heme is formed which irreversibly oxidizes "hotspot" residues (particularly βCys93) causing protein unfolding and subsequent heme loss. In this report we confirmed first, the allosteric effect of a newly developed reagent (di(5-(2,3-dihydro-1,4-benzodioxin-2-yl)-4H-1,2,4-triazol-3-yl)disulfide) (TD-1) on oxygen affinity within SS RBCs. There was a considerable left shift in oxygen equilibrium curves (OECs) representing treated SS cells. Under hypoxic conditions, TD-1 treatment of HbS resulted in an approximately 200 s increase in the delay time of HbS polymerization over the untreated HbS control. The effect of TD-1 binding to HbS was also tested on oxidative reactions by incrementally treating HbS with increasing hydrogen peroxide (HO) concentrations. Under these experimental conditions, ferryl levels were consistently reduced by approximately 35% in the presence of TD-1. Mass spectrometric analysis confirmed that upon binding to βCys93, TD-1 effectively blocked irreversible oxidation of this residue. In conclusion, TD-1 appears to shield βCys93 (the end point of radical formation in HbS) and when coupled with its allosteric effect on oxygen affinity may provide new therapeutic modalities for the treatment of SCD.
镰状细胞病(SCD)是一种遗传性血液疾病,由血红蛋白(HbS)的β球蛋白基因突变引起。脱氧 HbS 的聚合及其随后的聚集(形成长纤维)是导致红细胞(RBC)镰变并最终导致溶血性贫血的主要分子事件。我们最近提出,由于其缺陷的假过氧化物酶活性,HS 氧化毒性也可能导致 SCD 病理生理学。因此,形成了持续更高的氧化高铁血红素,其不可逆地氧化“热点”残基(特别是βCys93)导致蛋白质展开和随后的血红素损失。在本报告中,我们首先证实了一种新开发的试剂(二(5-(2,3-二氢-1,4-苯并二恶烷-2-基)-4H-1,2,4-三唑-3-基)二硫代)(TD-1)对 SS RBC 内氧亲和力的变构效应。氧平衡曲线(OEC)的左移相当大,代表治疗后的 SS 细胞。在缺氧条件下,HS 处理后,HS 聚合的延迟时间相对于未处理的 HbS 对照增加了约 200 秒。还通过逐步用增加的过氧化氢(HO)浓度处理 HbS 来测试 TD-1 与 HbS 结合对氧化反应的影响。在这些实验条件下,在存在 TD-1 的情况下,高铁血红素水平始终降低约 35%。质谱分析证实,TD-1 结合到βCys93 后,有效地阻止了该残基的不可逆氧化。总之,TD-1 似乎可以屏蔽βCys93(HS 中自由基形成的终点),并且当与氧亲和力的变构效应结合使用时,可能为 SCD 的治疗提供新的治疗方式。