Interdisciplinary Research Center on Biology and Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, China.
University of Chinese Academy of Sciences, Shanghai, China.
Aging Cell. 2017 Oct;16(5):1180-1190. doi: 10.1111/acel.12654. Epub 2017 Aug 7.
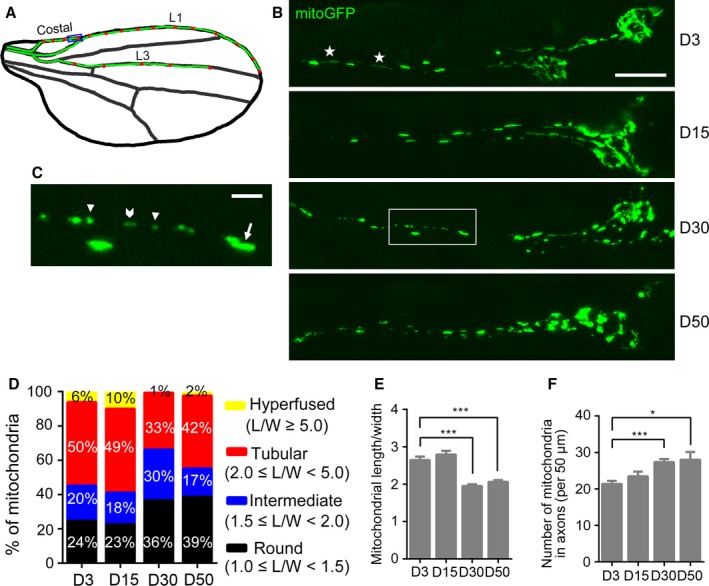
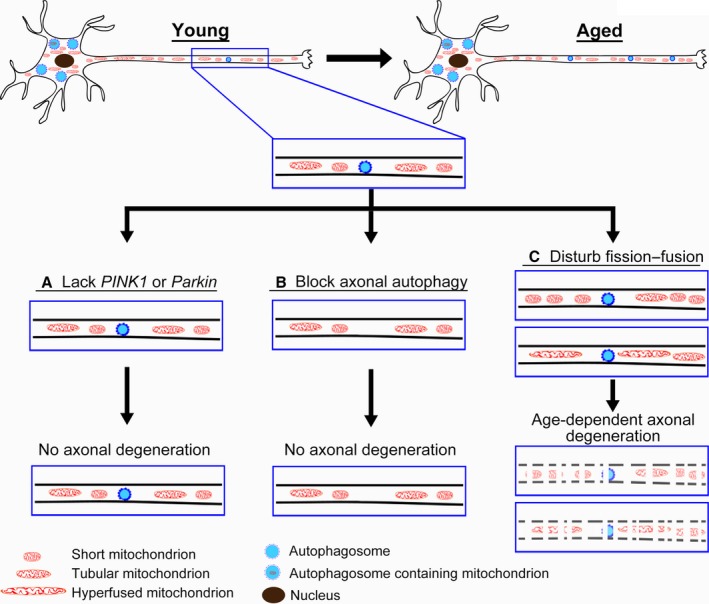
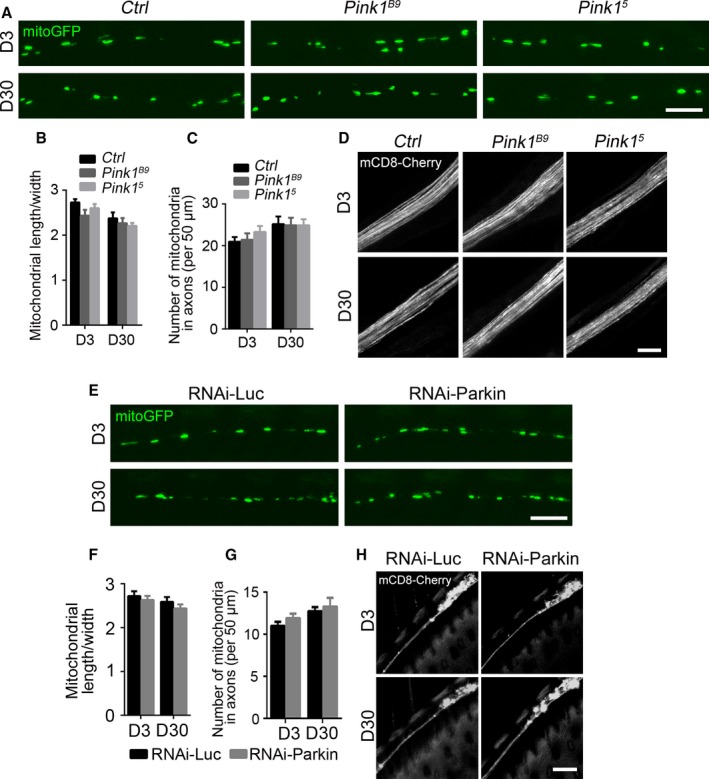
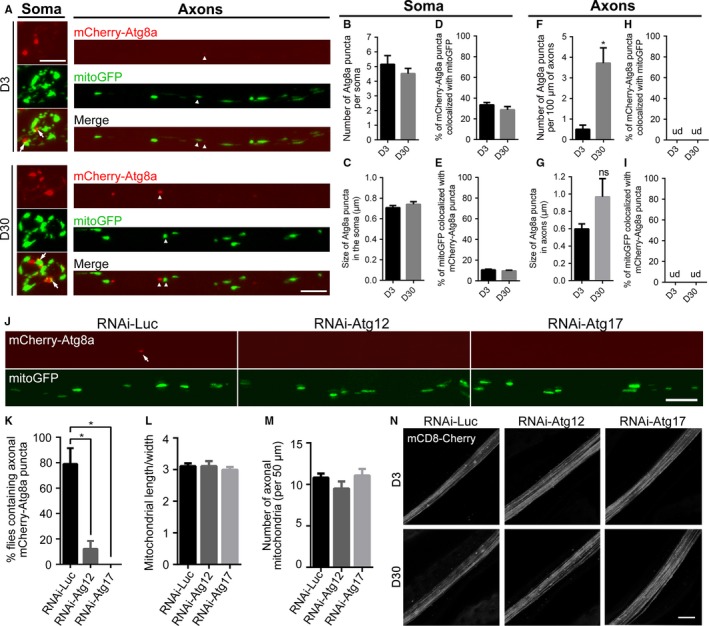
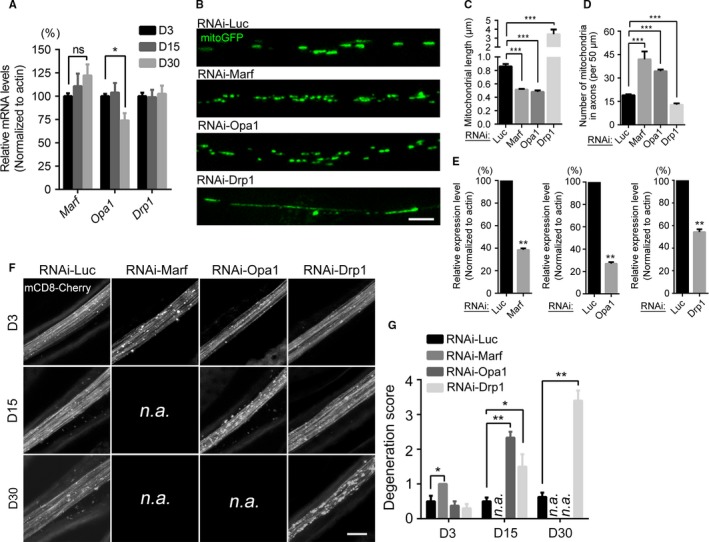
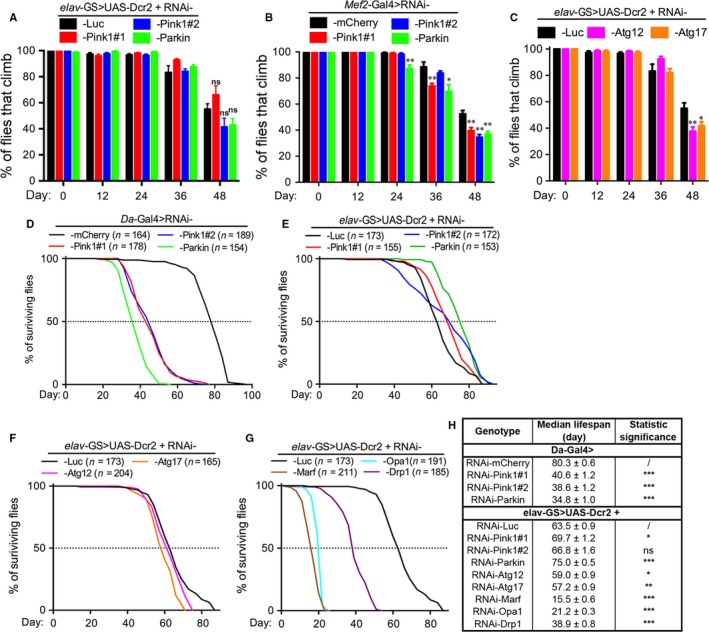
Mitophagy is thought to be a critical mitochondrial quality control mechanism in neurons and has been extensively studied in neurological disorders such as Parkinson's disease. However, little is known about how mitochondria are maintained in the lengthy neuronal axons in the context of physiological aging. Here, we utilized the unique Drosophila wing nerve model and in vivo imaging to rigorously profile changes in axonal mitochondria during aging. We revealed that mitochondria became fragmented and accumulated in aged axons. However, lack of Pink1 or Parkin did not lead to the accumulation of axonal mitochondria or axonal degeneration. Further, unlike in in vitro cultured neurons, we found that mitophagy rarely occurred in intact axons in vivo, even in aged animals. Furthermore, blocking overall mitophagy by knockdown of the core autophagy genes Atg12 or Atg17 had little effect on the turnover of axonal mitochondria or axonal integrity, suggesting that mitophagy is not required for axonal maintenance; this is regardless of whether the mitophagy is PINK1-Parkin dependent or independent. In contrast, downregulation of mitochondrial fission-fusion genes caused age-dependent axonal degeneration. Moreover, Opa1 expression in the fly head was significantly decreased with age, which may underlie the accumulation of fragmented mitochondria in aged axons. Finally, we showed that adult-onset, neuronal downregulation of the fission-fusion, but not mitophagy genes, dramatically accelerated features of aging. We propose that axonal mitochondria are maintained independently of mitophagy and that mitophagy-independent mechanisms such as fission-fusion may be central to the maintenance of axonal mitochondria and neural integrity during normal aging.
自噬被认为是神经元中线粒体质量控制的关键机制,在帕金森病等神经退行性疾病中得到了广泛研究。然而,在生理衰老的背景下,人们对神经元长轴突中线粒体的维持知之甚少。在这里,我们利用独特的果蝇翅神经模型和体内成像技术,严格分析了衰老过程中线粒体在轴突中的变化。结果显示,线粒体在衰老的轴突中变得碎片化并积累。然而,缺乏 Pink1 或 Parkin 并不会导致轴突中线粒体的积累或轴突退化。此外,与在体外培养的神经元不同,我们发现,在体内,即使在老年动物中,完整的轴突中很少发生自噬,即使在完整的轴突中也很少发生自噬。此外,通过敲低核心自噬基因 Atg12 或 Atg17 阻断整体自噬对轴突中线粒体的周转率或轴突完整性几乎没有影响,这表明自噬不是轴突维持所必需的;无论自噬是否依赖于 PINK1-Parkin。相比之下,下调线粒体分裂融合基因会导致年龄依赖性轴突退化。此外,果蝇头部的 Opa1 表达随年龄显著下降,这可能是衰老轴突中碎片化线粒体积累的原因。最后,我们发现,成虫后神经元下调分裂融合基因,但不影响自噬基因,会显著加速衰老特征。我们提出,轴突中线粒体的维持不依赖于自噬,而分裂融合等不依赖于自噬的机制可能是维持正常衰老过程中线粒体和神经完整性的关键。