Yue Weihua, Yu Xin, Zhang Dai
Institute of Mental Health, the Sixth Hospital, Peking University, 100191, Beijing, China.
Key Laboratory of Mental Health, Ministry of Health & National Clinical Research Center for Mental Disorders (Peking University), 100191, Beijing, China.
NPJ Schizophr. 2017 Aug 10;3(1):24. doi: 10.1038/s41537-017-0029-1.
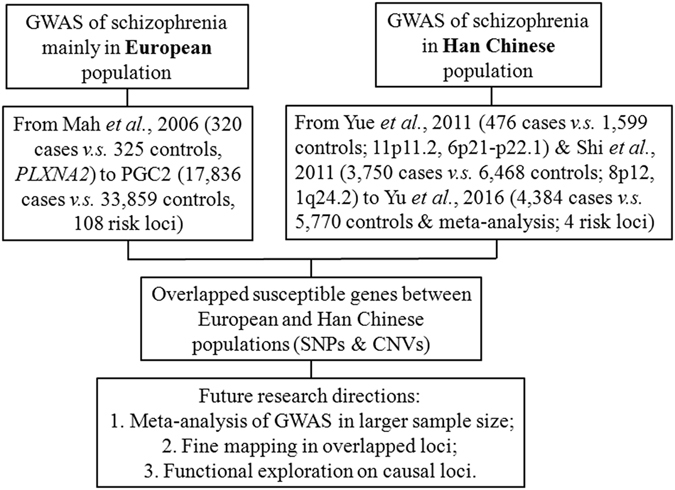
Since 2006, genome-wide association studies of schizophrenia have led to the identification of numerous novel risk loci for this disease. However, there remains a geographical imbalance in genome-wide association studies, which to date have primarily focused on Western populations. During the last 6 years, genome-wide association studies in Han Chinese populations have identified both the sharing of susceptible loci across ethnicities and genes unique to Han Chinese populations. Here, we review recent progress in genome-wide association studies of schizophrenia in Han Chinese populations. Researchers have identified and replicated the sharing of susceptible genes, such as within the major histocompatibility complex, microRNA 137 (MIR137), zinc finger protein 804A (ZNF804A), vaccinia related kinase 2 (VRK2), and arsenite methyltransferase (AS3MT), across both European and East Asian populations. Several copy number variations identified in European populations have also been validated in the Han Chinese, including duplications at 16p11.2, 15q11.2-13.1, 7q11.23, and VIPR2 and deletions at 22q11.2, 1q21.1-q21.2, and NRXN1. However, these studies have identified some potential confounding factors, such as genetic heterogeneity and the effects of natural selection on tetraspanin 18 (TSPAN18) or zinc finger protein 323 (ZNF323), which may explain the population differences in genome-wide association studies. In the future, genome-wide association studies in Han Chinese populations should include meta-analyzes or mega-analyses with enlarged sample sizes across populations, deep sequencing, precision medicine treatment, and functional exploration of the risk genes for schizophrenia.
自2006年以来,精神分裂症的全基因组关联研究已发现了该疾病的众多新风险基因座。然而,全基因组关联研究仍存在地域不平衡,迄今为止主要集中在西方人群。在过去6年中,针对汉族人群的全基因组关联研究既发现了不同种族间易感基因座的共享情况,也发现了汉族人群特有的基因。在此,我们综述汉族人群精神分裂症全基因组关联研究的最新进展。研究人员已经识别并重复验证了易感基因的共享情况,比如在欧洲和东亚人群中均存在的主要组织相容性复合体、微小RNA 137(MIR137)、锌指蛋白804A(ZNF804A)、痘苗相关激酶2(VRK2)和亚砷酸盐甲基转移酶(AS3MT)。在欧洲人群中识别出的几个拷贝数变异也在汉族人群中得到了验证,包括16p11.2、15q11.2 - 13.1、7q11.23和VIPR2的重复以及22q11.2、1q21.1 - q21.2和NRXN1的缺失。然而,这些研究也发现了一些潜在的混杂因素,如遗传异质性以及自然选择对四跨膜蛋白18(TSPAN18)或锌指蛋白323(ZNF323)的影响,这可能解释了全基因组关联研究中的人群差异。未来,汉族人群的全基因组关联研究应包括跨人群扩大样本量的荟萃分析或大型分析、深度测序、精准医学治疗以及对精神分裂症风险基因的功能探索。