Li Libei, Zhao Shuqi, Su Junji, Fan Shuli, Pang Chaoyou, Wei Hengling, Wang Hantao, Gu Lijiao, Zhang Chi, Liu Guoyuan, Yu Dingwei, Liu Qibao, Zhang Xianlong, Yu Shuxun
State Key Laboratory of Cotton Biology, Institute of Cotton Research of CAAS, Anyang, Henan, China.
National Key Laboratory of Crop Genetic Improvement, College of Plant Science and Technology, Huazhong Agricultural University, Wuhan, Hubei, China.
PLoS One. 2017 Aug 15;12(8):e0182918. doi: 10.1371/journal.pone.0182918. eCollection 2017.
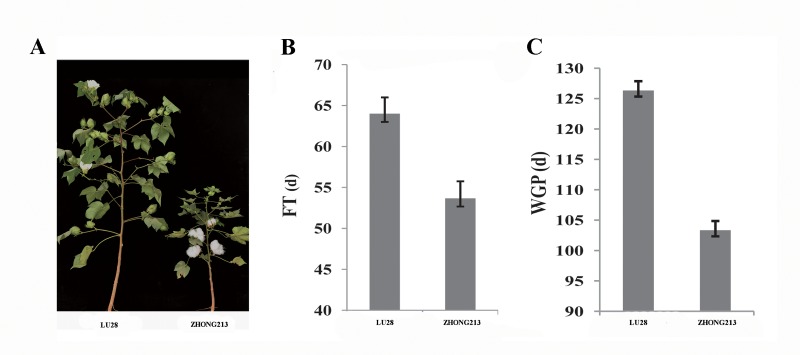
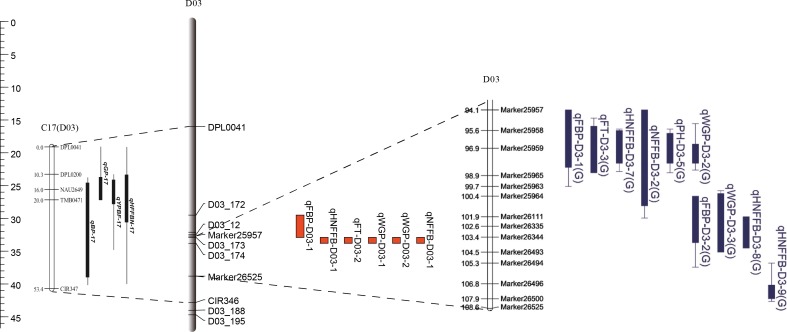
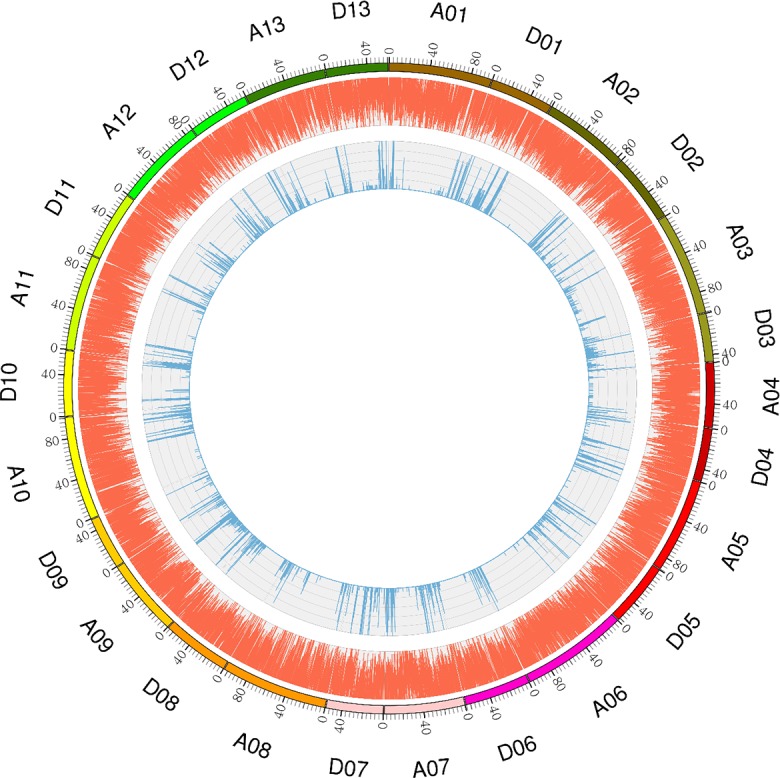
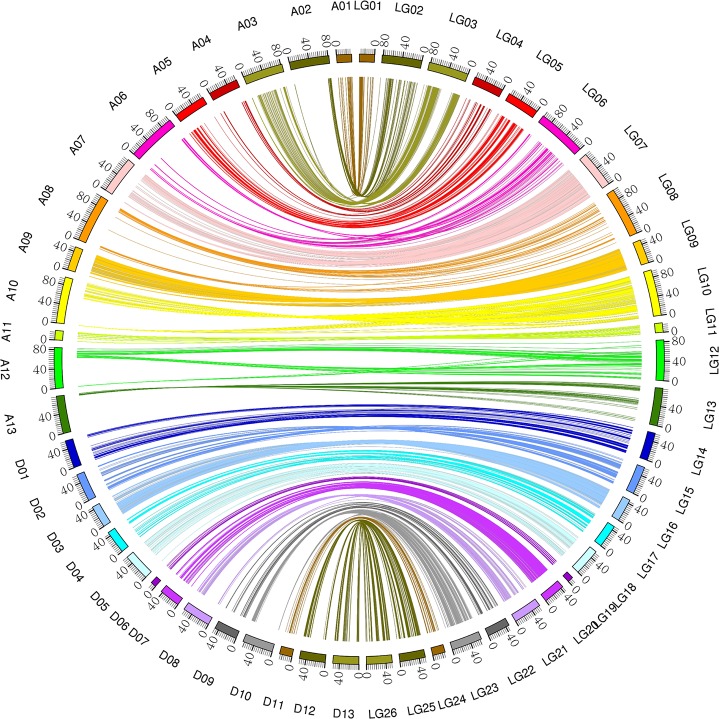
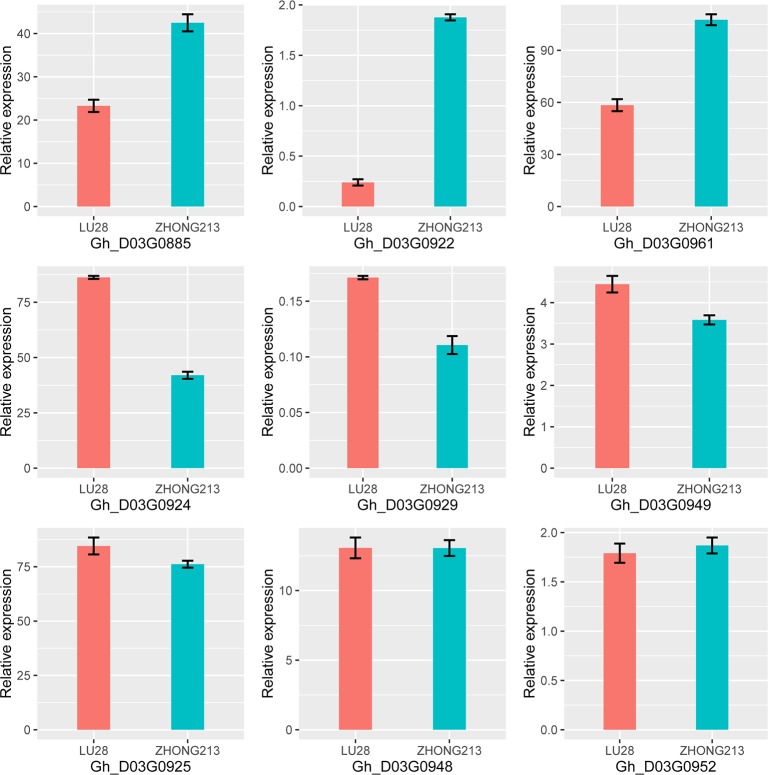
Due to China's rapidly increasing population, the total arable land area has dramatically decreased; as a consequence, the competition for farming land allocated for grain and cotton production has become fierce. Therefore, to overcome the existing contradiction between cotton grain and fiber production and the limited farming land, development of early-maturing cultivars is necessary. In this research, a high-density linkage map of upland cotton was constructed using genotyping by sequencing (GBS) to discover single nucleotide polymorphism (SNP) markers associated with early maturity in 170 F2 individuals derived from a cross between LU28 and ZHONG213. The high-density genetic map, which was composed of 3978 SNP markers across the 26 cotton chromosomes, spanned 2480 cM with an average genetic distance of 0.62 cM. Collinearity analysis showed that the genetic map was of high quality and accurate and agreed well with the Gossypium hirsutum reference genome. Based on this high-density linkage map, QTL analysis was performed on cotton early-maturity traits, including FT, FBP, WGP, NFFB, HNFFB and PH. A total 47 QTLs for the six traits were detected; each of these QTLs explained between 2.61% and 32.57% of the observed phenotypic variation. A major region controlling early-maturity traits in Gossypium hirsutum was identified for FT, FBP, WGP, NFFB and HNFFB on chromosome D03. QTL analyses revealed that phenotypic variation explained (PVE) ranged from 10.42% to 32.57%. Two potential candidate genes, Gh_D03G0885 and Gh_D03G0922, were predicted in a stable QTL region and had higher expression levels in the early-maturity variety ZHONG213 than in the late-maturity variety LU28. However, further evidence is required for functional validation. This study could provide useful information for the dissection of early-maturity traits and guide valuable genetic loci for molecular-assisted selection (MAS) in cotton breeding.
由于中国人口迅速增长,总耕地面积急剧减少;因此,用于粮食和棉花生产的耕地竞争变得激烈。所以,为了克服棉花粮食与纤维生产和有限耕地之间现有的矛盾,培育早熟品种是必要的。在本研究中,利用简化基因组测序(GBS)构建了陆地棉高密度连锁图谱,以发现与早熟相关的单核苷酸多态性(SNP)标记,该图谱来自LU28和中213杂交产生的170个F2个体。该高密度遗传图谱由分布在26条棉花染色体上的3978个SNP标记组成,跨度为2480厘摩,平均遗传距离为0.62厘摩。共线性分析表明,该遗传图谱质量高、准确性好,与陆地棉参考基因组吻合良好。基于此高密度连锁图谱,对棉花早熟性状进行了QTL分析,包括FT、FBP、WGP、NFFB、HNFFB和PH。共检测到这六个性状的47个QTL;每个QTL解释了2.61%至32.57%的表型变异。在D03染色体上鉴定出一个控制陆地棉早熟性状的主要区域,涉及FT、FBP、WGP、NFFB和HNFFB。QTL分析显示,表型变异解释率(PVE)在10.42%至32.57%之间。在一个稳定的QTL区域预测到两个潜在的候选基因Gh_D03G0885和Gh_D03G0922,它们在早熟品种中213中的表达水平高于晚熟品种LU28。然而,功能验证还需要进一步的证据。本研究可为解析早熟性状提供有用信息,并为棉花育种中的分子辅助选择(MAS)指导有价值的基因位点。