Albrecht-Kossel-Institute for Neuroregeneration (AKos), University Medicine Rostock, Gehlsheimer Straße 20, 18147, Rostock, Germany.
Orphanet J Rare Dis. 2017 Aug 25;12(1):145. doi: 10.1186/s13023-017-0697-y.
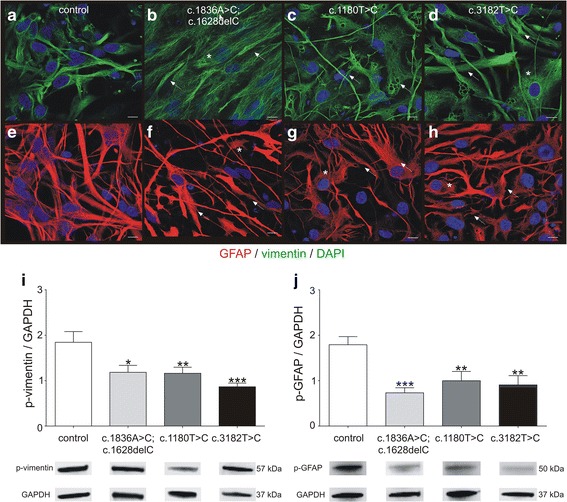
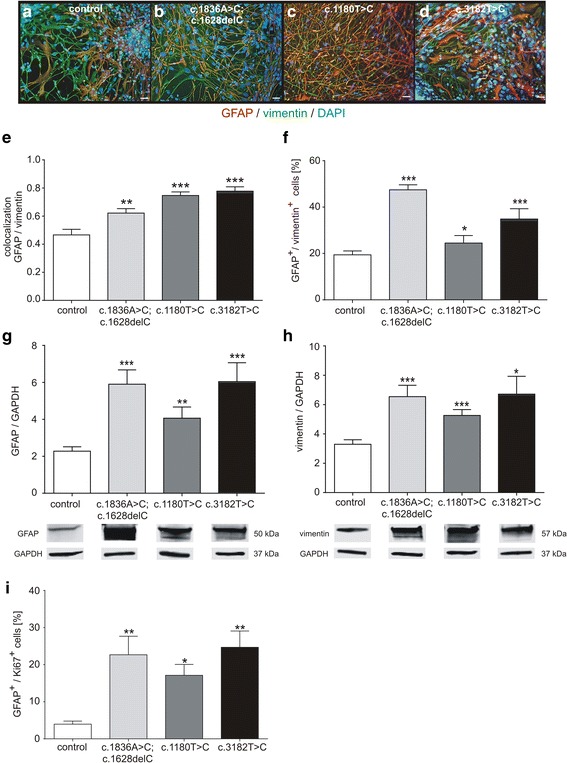
Niemann-Pick disease Type C1 (NPC1) is a rare progressive neurodegenerative disorder caused by mutations in the NPC1 gene. The pathological mechanisms, underlying NPC1 are not yet completely understood. Especially the contribution of glial cells and gliosis to the progression of NPC1, are controversially discussed. As an analysis of affected cells is unfeasible in NPC1-patients, we recently developed an in vitro model system, based on cells derived from NPC1-patient specific iPSCs. Here, we asked if this model system recapitulates gliosis, observed in non-human model systems and NPC1 patient post mortem biopsies. We determined the amount of reactive astrocytes and the regulation of the intermediate filaments GFAP and vimentin, all indicating gliosis. Furthermore, we were interested in the assembly and phosphorylation of these intermediate filaments and finally the impact of the activation of protein kinase C (PKC), which is described to ameliorate the pathogenic phenotype of NPC1-deficient fibroblasts, including hypo-phosphorylation of vimentin and cholesterol accumulation.
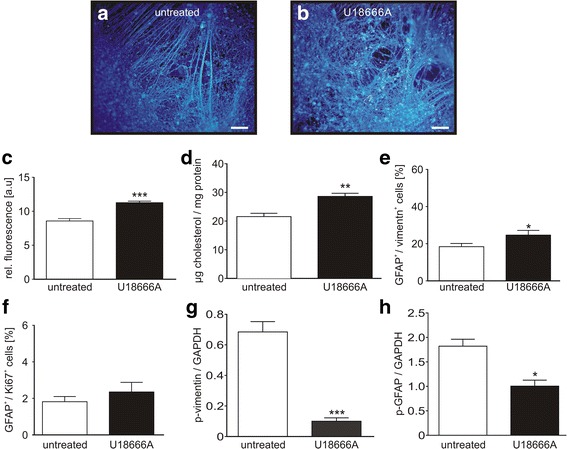
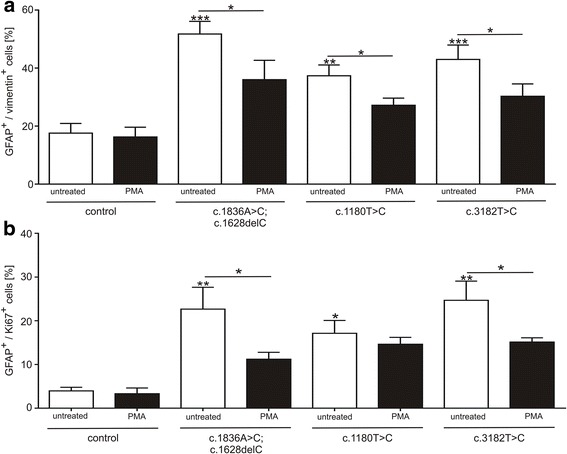
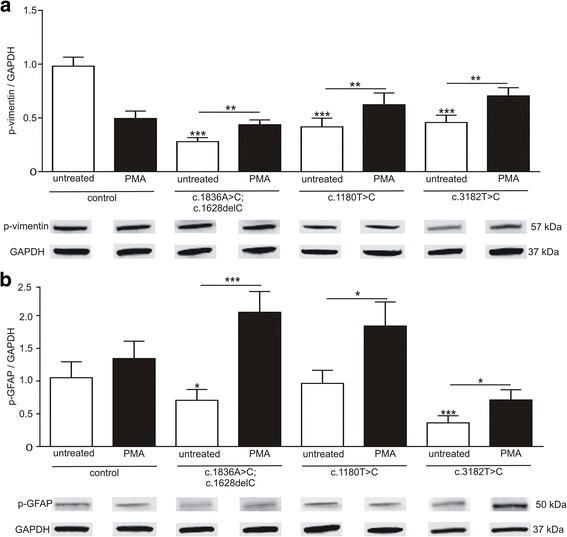
We analysed glial cells derived from NPC1 patient specific induced pluripotent stem cells, carrying different NPC1 mutations. The amount of reactive astrocytes was determined by means of immuncytochemical stainings and FACS-analysis. Semi-quantitative western blot was used to determine the amount of phosphorylated GFAP and vimentin. Cholesterol accumulation was analysed by Filipin staining and quantified by Amplex Red Assay. U18666A was used to induce NPC1 phenotype in unaffected cells of the control cell line. Phorbol 12-myristate 13-acetate (PMA) was used to activate PKC.
Immunocytochemical detection of GFAP, vimentin and Ki67 revealed that NPC1 mutant glial cells undergo gliosis. We found hypo-phosphorylation of the intermediate filaments GFAP and vimentin and alterations in the assembly of these intermediate filaments in NPC1 mutant cells. The application of U18666A induced not only NPC1 phenotypical accumulation of cholesterol, but characteristics of gliosis in glial cells derived from unaffected control cells. The application of phorbol 12-myristate 13-acetate, an activator of protein kinase C resulted in a significantly reduced number of reactive astrocytes and further characteristics of gliosis in NPC1-deficient cells. Furthermore, it triggered a restoration of cholesterol amounts to level of control cells.
Our data demonstrate that glial cells derived from NPC1-patient specific iPSCs undergo gliosis. The application of U18666A induced comparable characteristics in un-affected control cells, suggesting that gliosis is triggered by hampered function of NPC1 protein. The activation of protein kinase C induced an amelioration of gliosis, as well as a reduction of cholesterol amount. These results provide further support for the line of evidence that gliosis might not be only a secondary reaction to the loss of neurons, but might be a direct consequence of a reduced PKC activity due to the phenotypical cholesterol accumulation observed in NPC1. In addition, our data support the involvement of PKCs in NPC1 disease pathogenesis and suggest that PKCs may be targeted in future efforts to develop therapeutics for NPC1 disease.
尼曼-皮克病 C1 型(NPC1)是一种由 NPC1 基因突变引起的罕见进行性神经退行性疾病。NPC1 的病理机制尚未完全清楚。特别是神经胶质细胞和神经胶质增生对 NPC1 进展的贡献存在争议。由于无法对 NPC1 患者的受影响细胞进行分析,我们最近基于 NPC1 患者特异性诱导多能干细胞(iPSC)开发了一种体外模型系统。在这里,我们询问该模型系统是否能再现非人类模型系统和 NPC1 患者死后活检中观察到的神经胶质增生。我们确定了反应性星形胶质细胞的数量以及中间丝 GFAP 和波形蛋白的调节,所有这些都表明存在神经胶质增生。此外,我们还对这些中间丝的组装和磷酸化以及蛋白激酶 C(PKC)的激活感兴趣,PKC 的激活被描述为改善 NPC1 缺陷成纤维细胞的致病表型,包括波形蛋白的低磷酸化和胆固醇积累。
我们分析了来自携带不同 NPC1 突变的 NPC1 患者特异性诱导多能干细胞的神经胶质细胞。通过免疫细胞化学染色和流式细胞术分析确定反应性星形胶质细胞的数量。半定量 Western blot 用于确定磷酸化 GFAP 和波形蛋白的量。用 Filipin 染色分析胆固醇积累,并通过 Amplex Red 测定法进行定量。U18666A 用于诱导对照细胞系中未受影响细胞的 NPC1 表型。佛波醇 12-肉豆蔻酸 13-乙酸酯(PMA)用于激活 PKC。
GFAP、波形蛋白和 Ki67 的免疫细胞化学检测表明 NPC1 突变的神经胶质细胞发生神经胶质增生。我们发现 NPC1 突变细胞中的中间丝 GFAP 和波形蛋白的低磷酸化以及这些中间丝的组装发生改变。U18666A 的应用不仅诱导了 NPC1 表型胆固醇的积累,而且还诱导了源自未受影响对照细胞的神经胶质细胞的神经胶质增生特征。蛋白激酶 C 的激活剂佛波醇 12-肉豆蔻酸 13-乙酸酯(PMA)的应用导致 NPC1 缺陷细胞中反应性星形胶质细胞数量显著减少,并进一步表现出神经胶质增生的特征。此外,它还触发了胆固醇水平恢复到对照细胞的水平。
我们的数据表明,源自 NPC1 患者特异性 iPSC 的神经胶质细胞发生神经胶质增生。U18666A 的应用在未受影响的对照细胞中诱导了类似的特征,表明神经胶质增生是 NPC1 蛋白功能障碍引起的。蛋白激酶 C 的激活诱导了神经胶质增生的改善以及胆固醇含量的减少。这些结果进一步支持了这样的证据,即神经胶质增生不仅可能是神经元丢失的继发反应,而且可能是 NPC1 中观察到的表型胆固醇积累导致 PKC 活性降低的直接后果。此外,我们的数据支持 PKC 参与 NPC1 疾病发病机制,并表明 PKC 可能成为未来开发 NPC1 疾病治疗方法的靶点。