Kormann Michael S D, Dewerth Alexander, Eichner Felizitas, Baskaran Praveen, Hector Andreas, Regamey Nicolas, Hartl Dominik, Handgretinger Rupert, Antony Justin S
Department of Pediatrics I, Pediatric Infectiology and Immunology, Translational Genomics and Gene Therapy in Pediatrics, University of Tuebingen, Tuebingen, Germany.
Center for Quantitative Biology, University of Tuebingen, Tuebingen, Germany.
PLoS One. 2017 Aug 28;12(8):e0183526. doi: 10.1371/journal.pone.0183526. eCollection 2017.
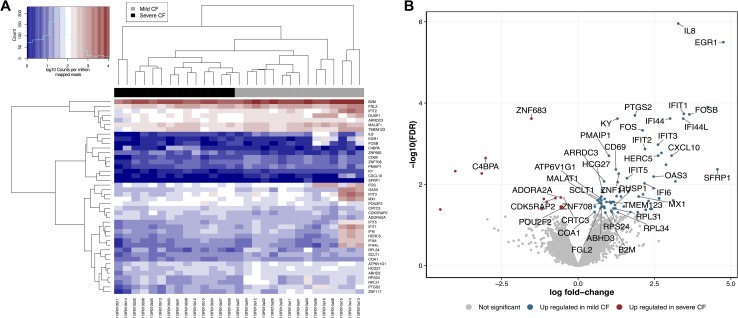
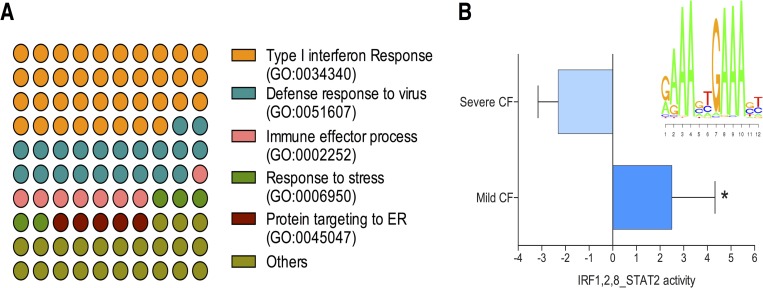
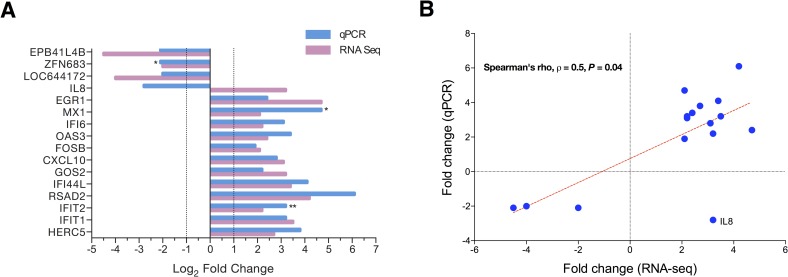
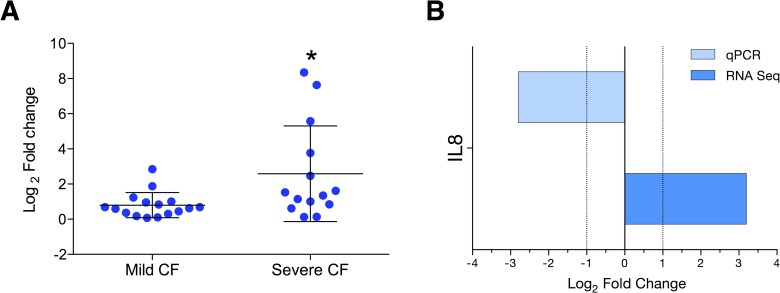
Cystic Fibrosis (CF) is the most common monogenic disease among people of Western European descent and caused by mutations in the CFTR gene. However, the disease severity is immensely variable even among patients with similar CFTR mutations due to the possible effect of 'modifier genes'. To identify genetic modifiers, we applied RNA-seq based transcriptomic analyses in CF patients with a mild and severe lung phenotype. Global gene expression and enrichment analyses revealed that genes of the type I interferon response and ribosomal stalk proteins are potential modifiers of CF related lung dysfunction. The results provide a new set of CF modifier genes with possible implications as new therapeutic targets for the treatment of CF.
囊性纤维化(CF)是西欧血统人群中最常见的单基因疾病,由CFTR基因突变引起。然而,由于“修饰基因”的可能影响,即使在具有相似CFTR突变的患者中,疾病严重程度也存在极大差异。为了鉴定遗传修饰因子,我们对具有轻度和重度肺部表型的CF患者进行了基于RNA测序的转录组分析。整体基因表达和富集分析表明,I型干扰素反应基因和核糖体柄蛋白是CF相关肺功能障碍的潜在修饰因子。这些结果提供了一组新的CF修饰基因,可能作为CF治疗的新治疗靶点具有重要意义。