SIB Swiss Institute of Bioinformatics, Basel, Switzerland.
Biozentrum, University of Basel, Klingelbergstrasse 50/70, 4056, Basel, Switzerland.
Sci Rep. 2017 Sep 5;7(1):10480. doi: 10.1038/s41598-017-09654-8.
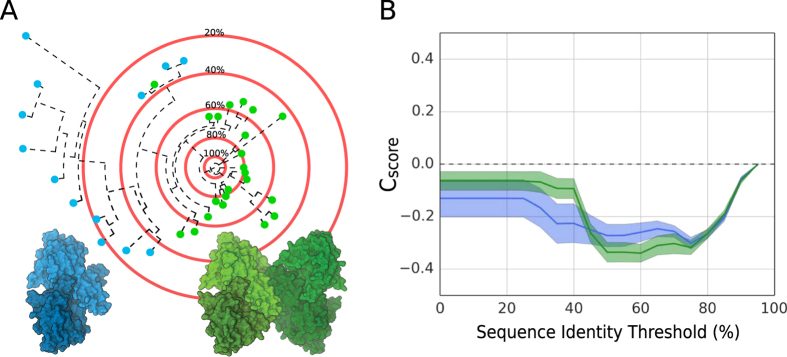
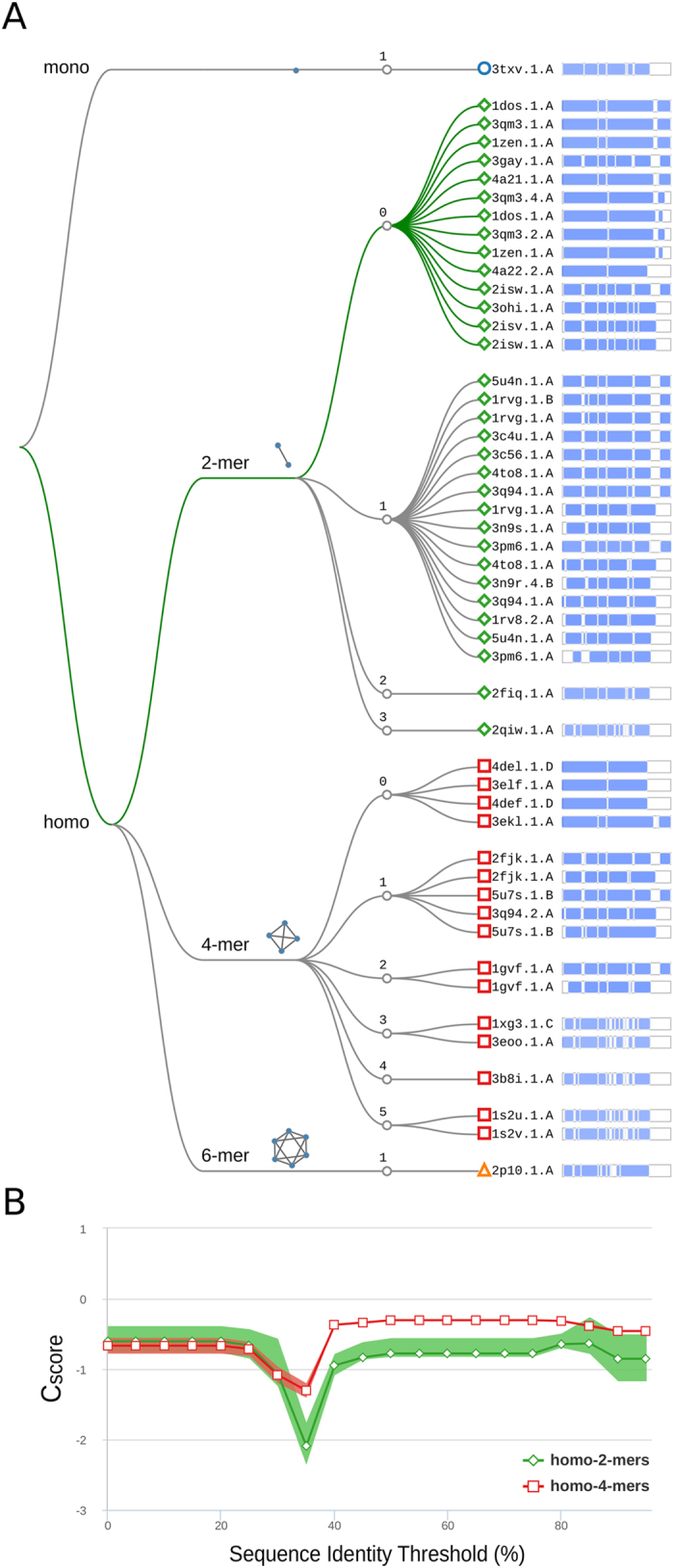
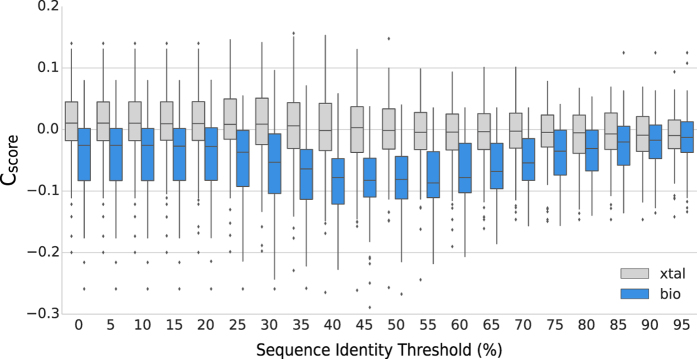
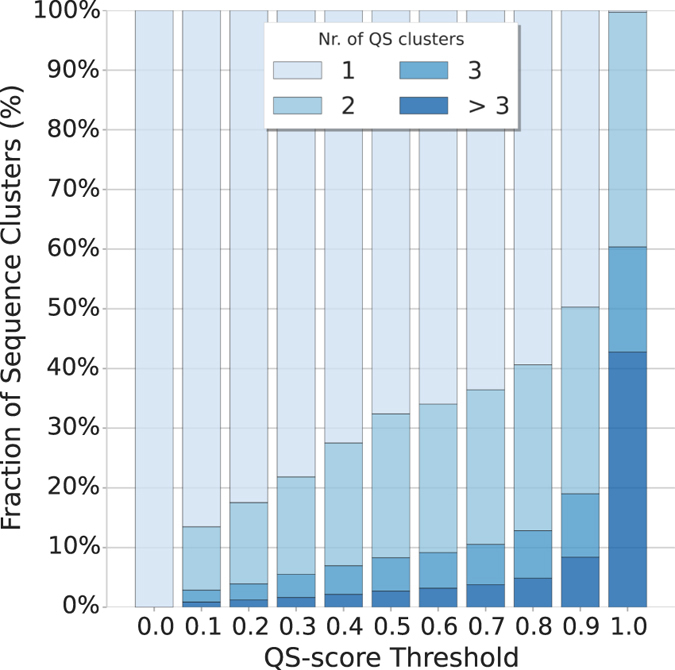
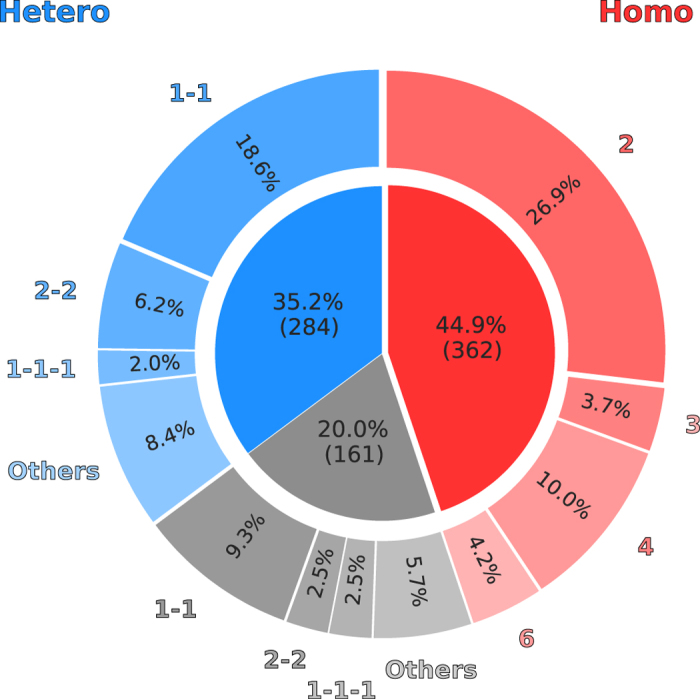
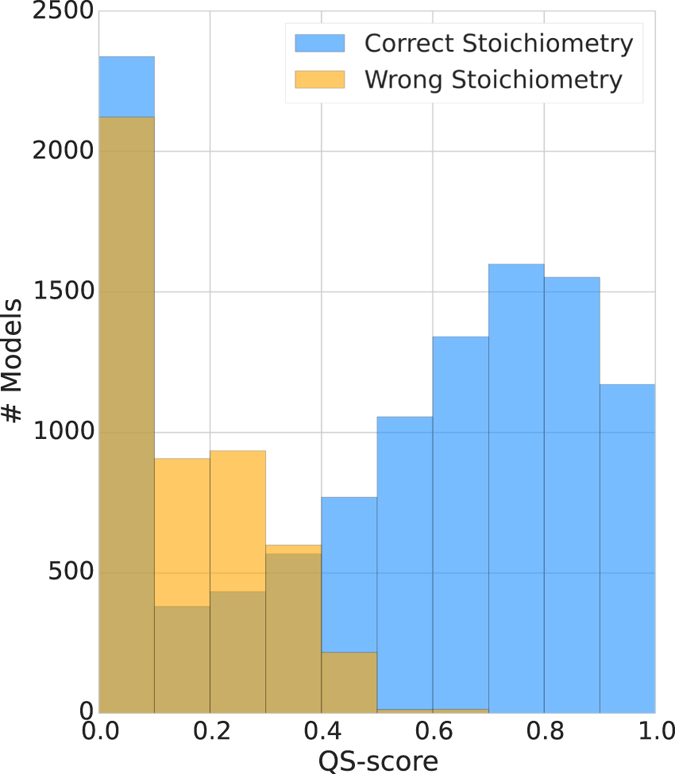
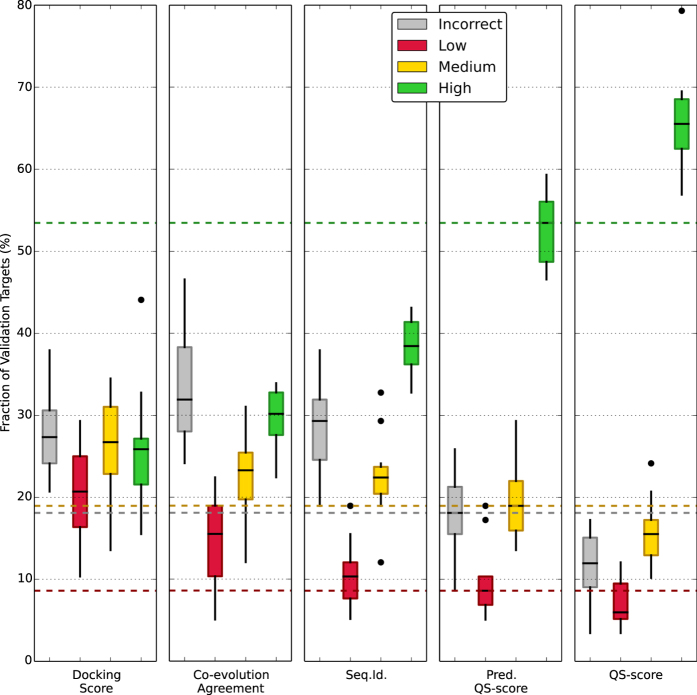
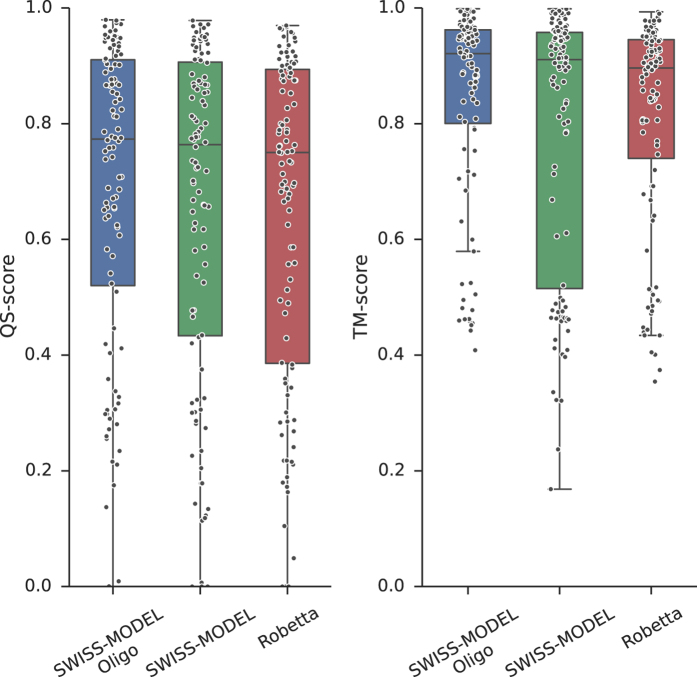
Cellular processes often depend on interactions between proteins and the formation of macromolecular complexes. The impairment of such interactions can lead to deregulation of pathways resulting in disease states, and it is hence crucial to gain insights into the nature of macromolecular assemblies. Detailed structural knowledge about complexes and protein-protein interactions is growing, but experimentally determined three-dimensional multimeric assemblies are outnumbered by complexes supported by non-structural experimental evidence. Here, we aim to fill this gap by modeling multimeric structures by homology, only using amino acid sequences to infer the stoichiometry and the overall structure of the assembly. We ask which properties of proteins within a family can assist in the prediction of correct quaternary structure. Specifically, we introduce a description of protein-protein interface conservation as a function of evolutionary distance to reduce the noise in deep multiple sequence alignments. We also define a distance measure to structurally compare homologous multimeric protein complexes. This allows us to hierarchically cluster protein structures and quantify the diversity of alternative biological assemblies known today. We find that a combination of conservation scores, structural clustering, and classical interface descriptors, can improve the selection of homologous protein templates leading to reliable models of protein complexes.
细胞过程通常依赖于蛋白质之间的相互作用和形成的大分子复合物。这种相互作用的损害会导致途径失调,导致疾病状态,因此深入了解大分子组装的性质至关重要。关于复合物和蛋白质-蛋白质相互作用的详细结构知识在不断增加,但实验确定的三维多聚体组装数量超过了仅由非结构实验证据支持的复合物。在这里,我们旨在通过同源建模来填补这一空白,仅使用氨基酸序列来推断组装的化学计量和整体结构。我们询问蛋白质家族中的哪些特性可以帮助预测正确的四级结构。具体来说,我们引入了一种蛋白质-蛋白质界面保守性的描述,作为进化距离的函数,以减少深度多重序列比对中的噪声。我们还定义了一种距离度量标准来结构比较同源多聚体蛋白复合物。这使我们能够对蛋白质结构进行层次聚类,并量化当今已知的替代生物组装的多样性。我们发现,保守分数、结构聚类和经典界面描述符的组合,可以改进同源蛋白模板的选择,从而得到可靠的蛋白复合物模型。