Lorillon Gwenaël, Tazi Abdellatif
National Reference Centre for Histiocytoses, Pulmonary Dept, Assistance Publique-Hôpitaux de Paris, Hôpital Saint-Louis, Paris, France.
National Reference Centre for Histiocytoses, Pulmonary Dept, Assistance Publique-Hôpitaux de Paris, Hôpital Saint-Louis, Paris, France
Eur Respir Rev. 2017 Sep 6;26(145). doi: 10.1183/16000617.0070-2017. Print 2017 Sep 30.
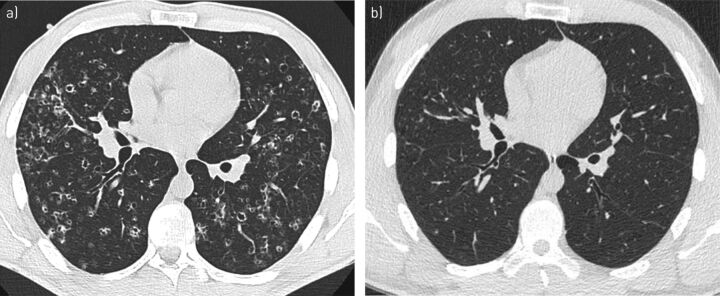
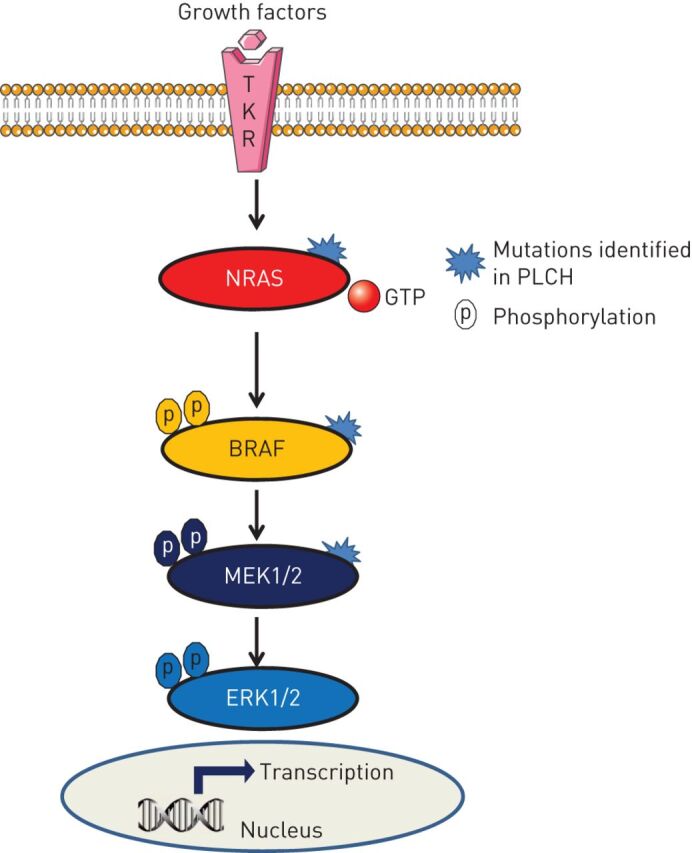
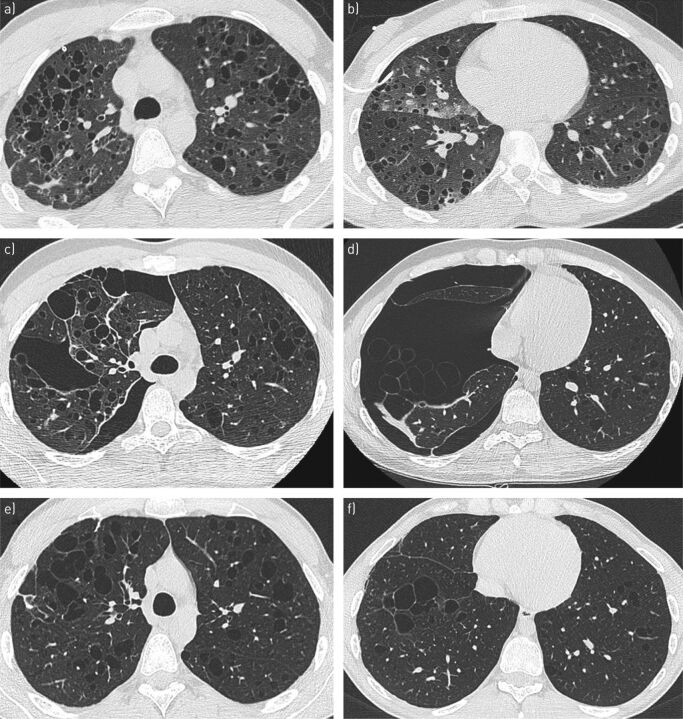
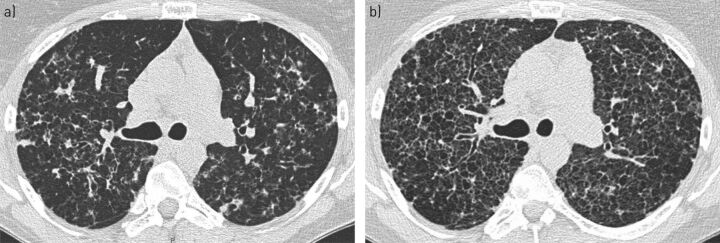
Pulmonary Langerhans cell histiocytosis (PLCH) is a rare sporadic cystic lung disease of unknown aetiology that is characterised by the infiltration and destruction of the wall of distal bronchioles by CD1a Langerhans-like cells. In adults, PLCH is frequently isolated and affects young smokers of both sexes. Recent multicentre studies have led to the more standardised management of patients in clinical practice. Smoking cessation is essential and is occasionally the only suitable intervention. Serial lung function testing is important because a significant proportion of patients may experience an early decline in forced expiratory volume in 1 s and develop airflow obstruction. Cladribine was reported to dramatically improve progressive PLCH in some patients. Its efficacy and tolerance are currently being evaluated. Patients who complain of unexplained dyspnoea with decreased diffusing capacity of the lung for carbon monoxide should be screened for pulmonary hypertension by Doppler echocardiography, which must be confirmed by right heart catheterisation. Lung transplantation is a therapeutic option for patients with advanced PLCH.The identification of the mutation in approximately half of Langerhans cell histiocytosis lesions, including PLCH, and other mutations of the mitogen-activated protein kinase (MAPK) pathway in a subset of lesions has led to targeted treatments (BRAF and MEK (MAPK kinase) inhibitors). These treatments need to be rigorously evaluated because of their potentially severe side-effects.
肺朗格汉斯细胞组织细胞增多症(PLCH)是一种病因不明的罕见散发性囊性肺部疾病,其特征是CD1a朗格汉斯样细胞浸润并破坏终末细支气管壁。在成年人中,PLCH通常为孤立性,且男女青年吸烟者均会受累。最近的多中心研究使临床实践中患者的管理更加标准化。戒烟至关重要,有时是唯一合适的干预措施。定期进行肺功能测试很重要,因为相当一部分患者可能会出现1秒用力呼气量早期下降并发展为气流受限。据报道,克拉屈滨可使部分患者的进行性PLCH显著改善。目前正在评估其疗效和耐受性。对于主诉不明原因呼吸困难且肺一氧化碳弥散能力下降的患者,应通过多普勒超声心动图筛查肺动脉高压,必须通过右心导管检查予以确诊。肺移植是晚期PLCH患者的一种治疗选择。在大约一半的朗格汉斯细胞组织细胞增多症病变(包括PLCH)中发现了该突变,以及在一部分病变中发现了丝裂原活化蛋白激酶(MAPK)途径的其他突变,从而带来了靶向治疗(BRAF和MEK(MAPK激酶)抑制剂)。由于这些治疗可能有严重的副作用,因此需要进行严格评估。