Hücker Sarah M, Ardern Zachary, Goldberg Tatyana, Schafferhans Andrea, Bernhofer Michael, Vestergaard Gisle, Nelson Chase W, Schloter Michael, Rost Burkhard, Scherer Siegfried, Neuhaus Klaus
Chair for Microbial Ecology, Technische Universität München, Freising, Germany.
ZIEL - Institute for Food & Health, Technische Universität München, Freising, Germany.
PLoS One. 2017 Sep 13;12(9):e0184119. doi: 10.1371/journal.pone.0184119. eCollection 2017.
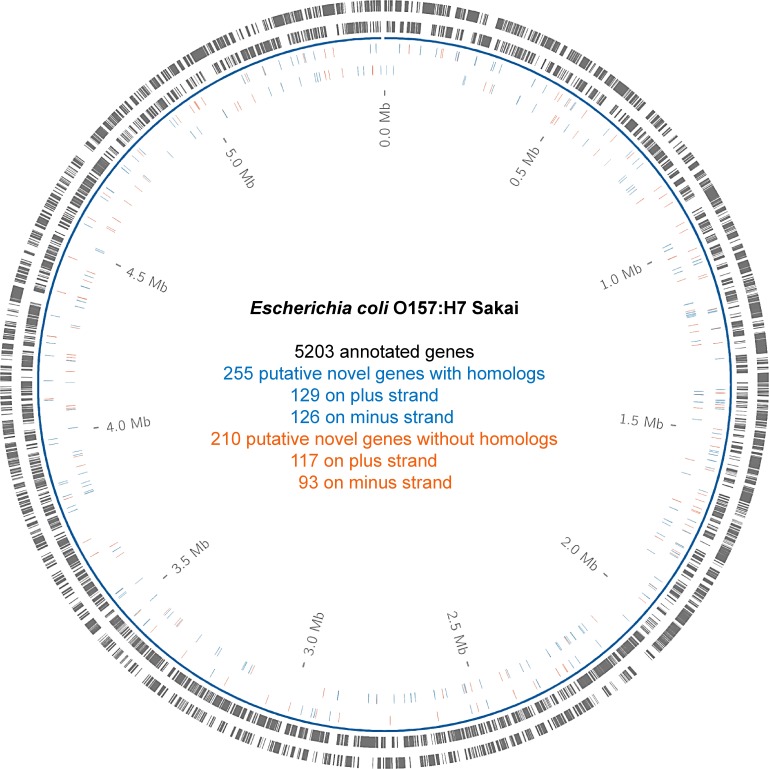
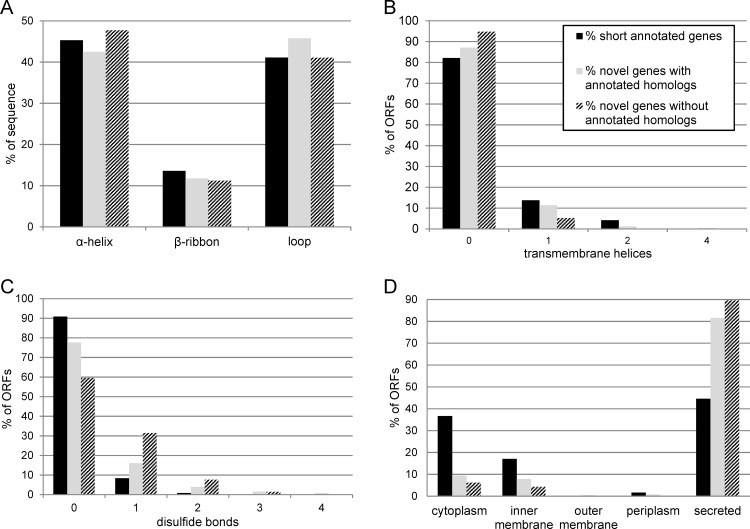
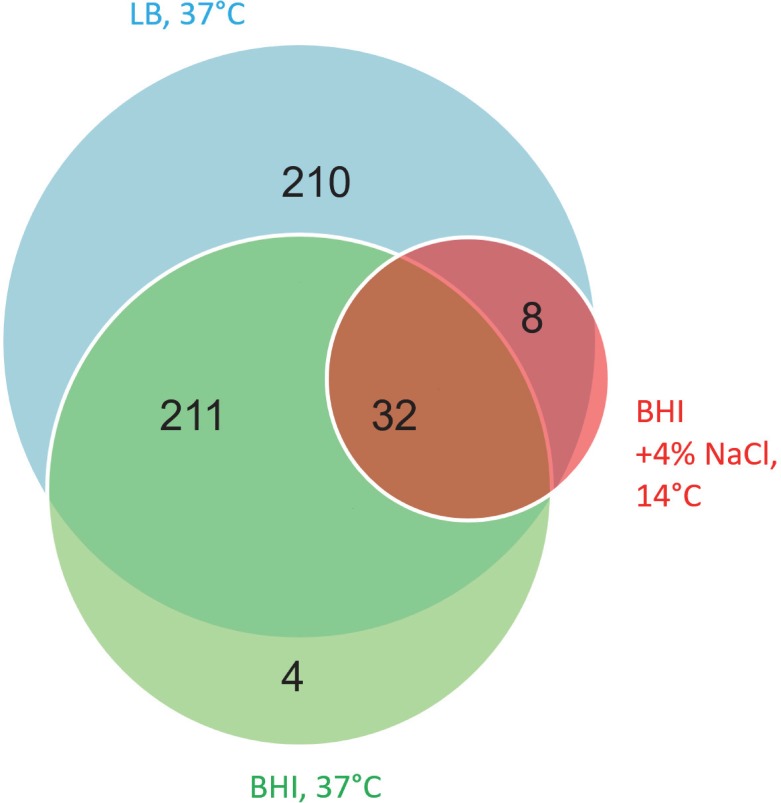
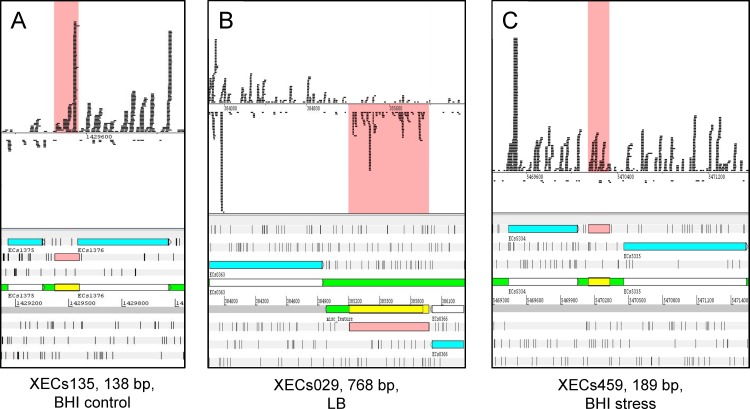
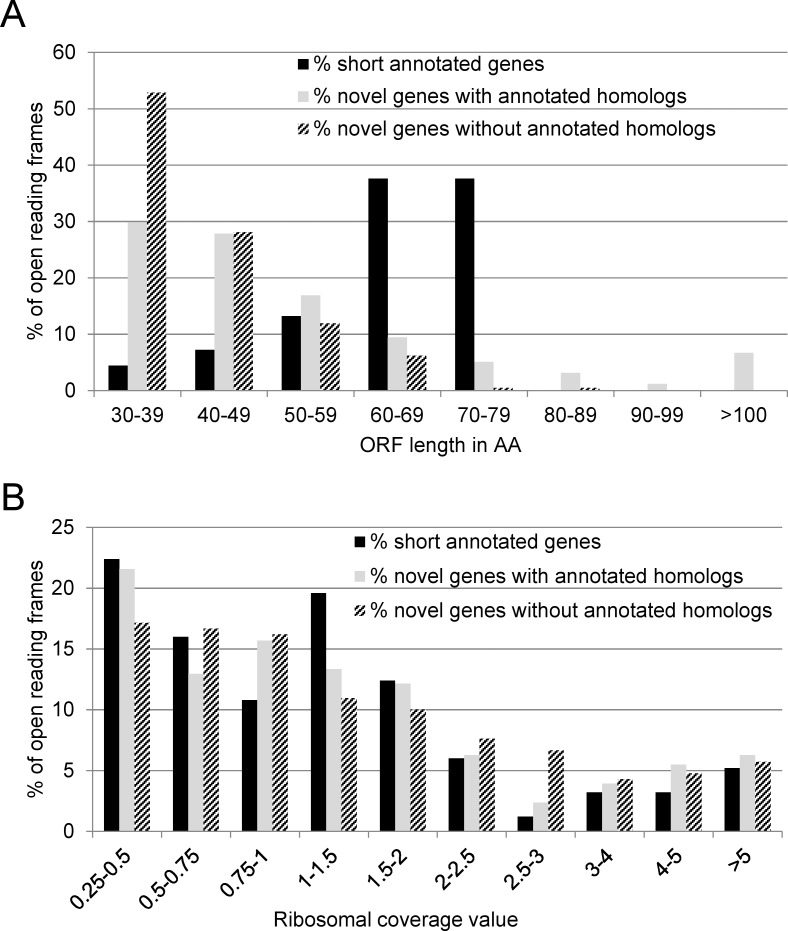
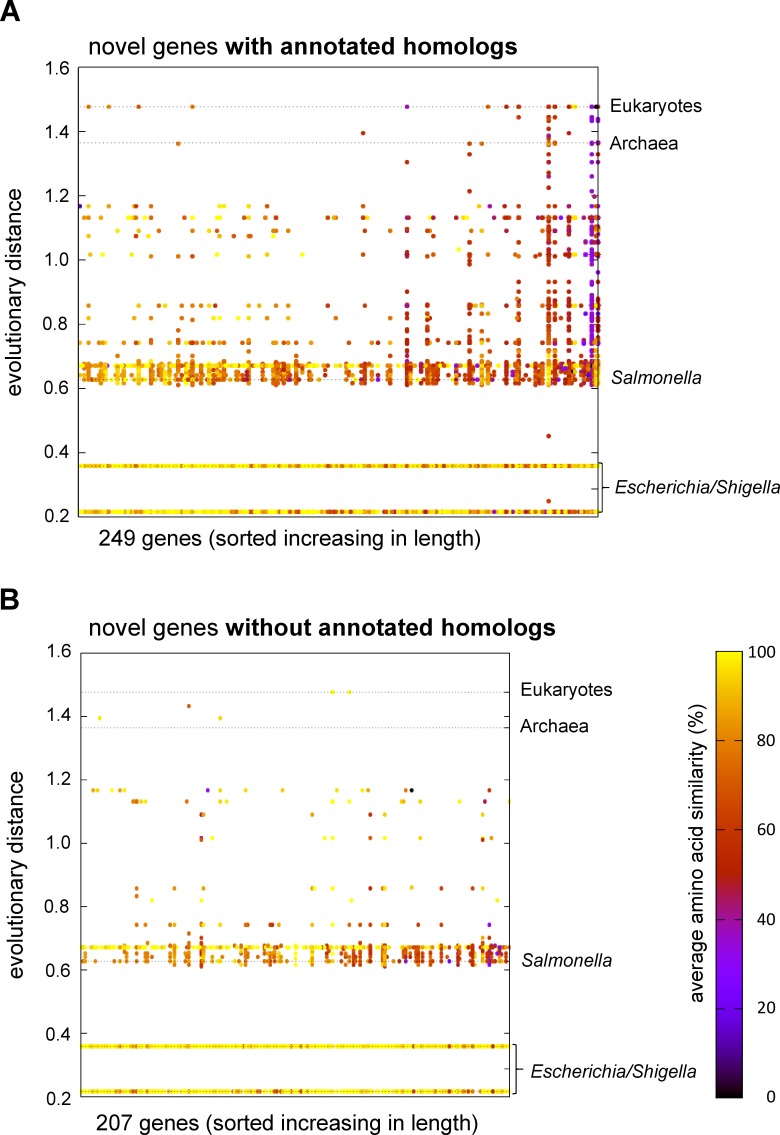
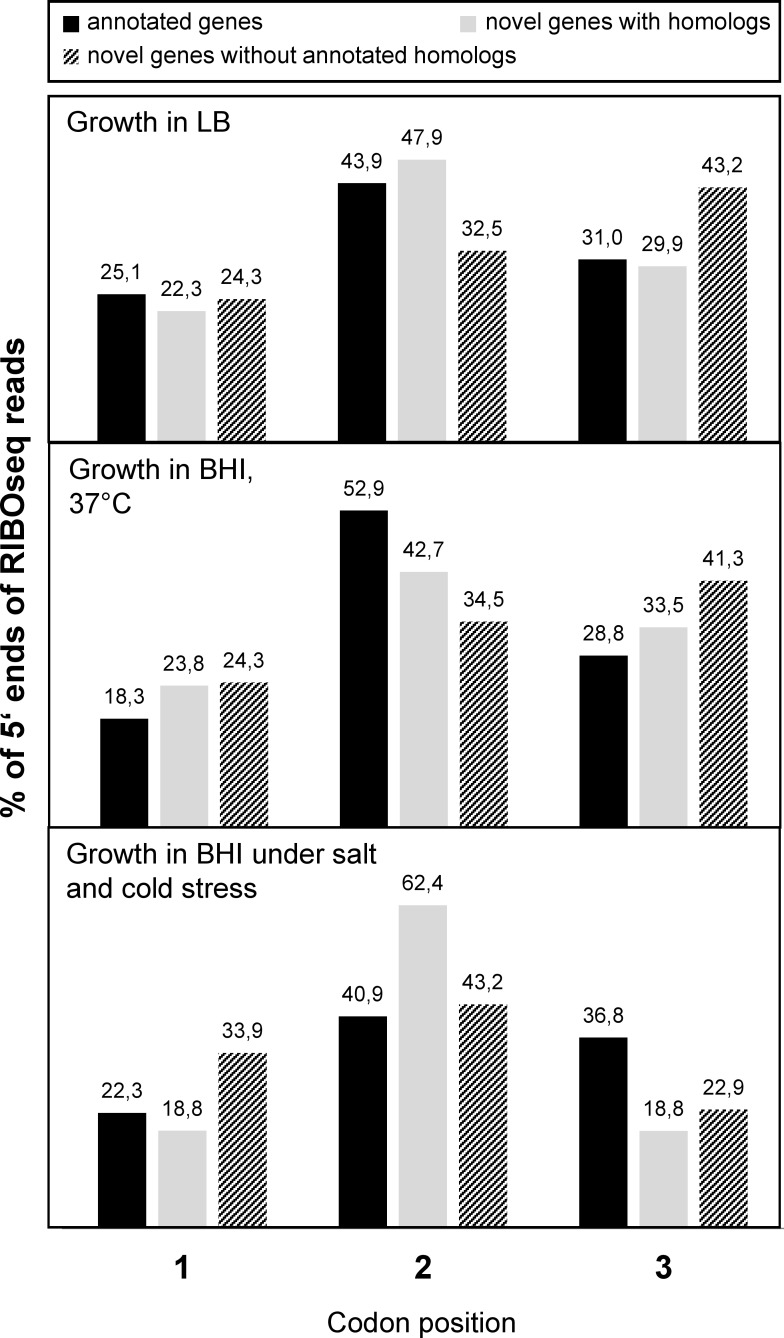
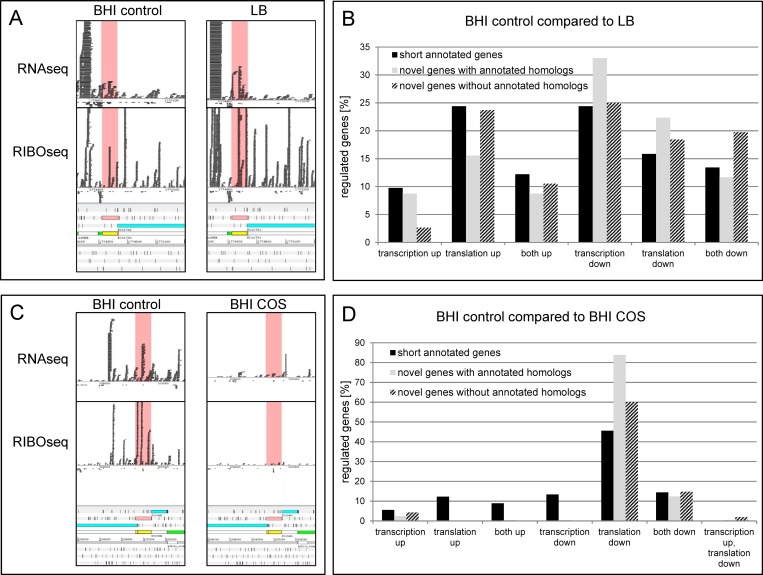
In the past, short protein-coding genes were often disregarded by genome annotation pipelines. Transcriptome sequencing (RNAseq) signals outside of annotated genes have usually been interpreted to indicate either ncRNA or pervasive transcription. Therefore, in addition to the transcriptome, the translatome (RIBOseq) of the enteric pathogen Escherichia coli O157:H7 strain Sakai was determined at two optimal growth conditions and a severe stress condition combining low temperature and high osmotic pressure. All intergenic open reading frames potentially encoding a protein of ≥ 30 amino acids were investigated with regard to coverage by transcription and translation signals and their translatability expressed by the ribosomal coverage value. This led to discovery of 465 unique, putative novel genes not yet annotated in this E. coli strain, which are evenly distributed over both DNA strands of the genome. For 255 of the novel genes, annotated homologs in other bacteria were found, and a machine-learning algorithm, trained on small protein-coding E. coli genes, predicted that 89% of these translated open reading frames represent bona fide genes. The remaining 210 putative novel genes without annotated homologs were compared to the 255 novel genes with homologs and to 250 short annotated genes of this E. coli strain. All three groups turned out to be similar with respect to their translatability distribution, fractions of differentially regulated genes, secondary structure composition, and the distribution of evolutionary constraint, suggesting that both novel groups represent legitimate genes. However, the machine-learning algorithm only recognized a small fraction of the 210 genes without annotated homologs. It is possible that these genes represent a novel group of genes, which have unusual features dissimilar to the genes of the machine-learning algorithm training set.
过去,短的蛋白质编码基因常常被基因组注释流程所忽视。注释基因之外的转录组测序(RNAseq)信号通常被解释为表明非编码RNA(ncRNA)或广泛转录。因此,除了转录组,还在两种最佳生长条件以及低温和高渗透压相结合的严重应激条件下,测定了肠道病原体大肠杆菌O157:H7菌株阪崎的翻译组(核糖体印迹测序,RIBOseq)。研究了所有可能编码≥30个氨基酸的蛋白质的基因间开放阅读框,涉及转录和翻译信号的覆盖情况以及由核糖体覆盖值表示的它们的可翻译性。这导致发现了465个在该大肠杆菌菌株中尚未注释的独特的、假定的新基因,它们均匀分布在基因组的两条DNA链上。对于其中255个新基因,在其他细菌中发现了注释的同源物,并且一种基于小的大肠杆菌蛋白质编码基因训练的机器学习算法预测,这些翻译后的开放阅读框中有89%代表真正的基因。将其余210个没有注释同源物的假定新基因与255个有同源物的新基因以及该大肠杆菌菌株的250个短注释基因进行了比较。结果发现,所有这三组在可翻译性分布、差异调节基因的比例、二级结构组成以及进化约束分布方面都相似,这表明这两个新组都代表合理的基因。然而,机器学习算法只识别出了210个没有注释同源物的基因中的一小部分。有可能这些基因代表了一组具有与机器学习算法训练集基因不同寻常特征的新基因。