Mulliken Center for Theoretical Chemistry, Institut für Physikalische und Theoretische Chemie der Universität Bonn, Beringstrasse 4, 53115, Bonn, Germany.
Max Planck Institute for Chemical Energy Conversion, Stiftstrasse 32-34, 45470, Mülheim an der Ruhr, Germany.
Angew Chem Int Ed Engl. 2017 Nov 13;56(46):14763-14769. doi: 10.1002/anie.201708266. Epub 2017 Oct 11.
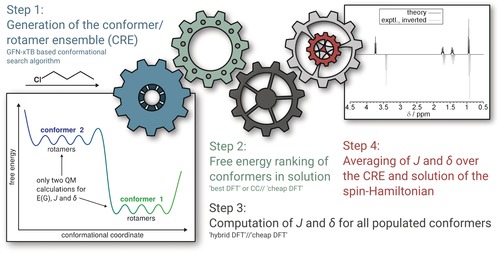
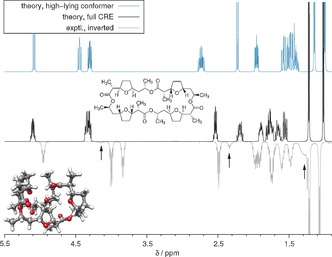
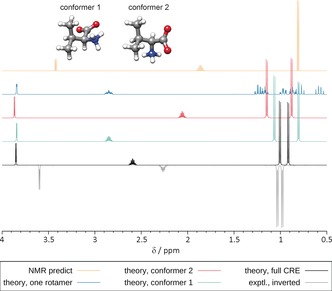
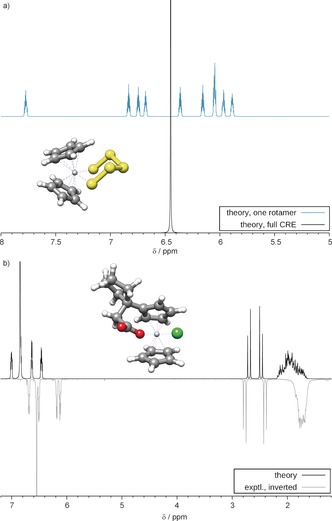
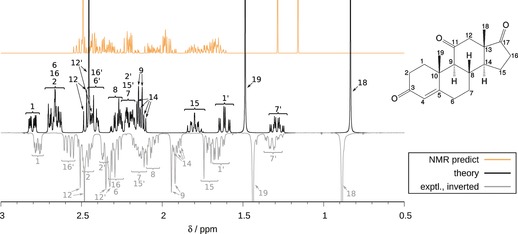
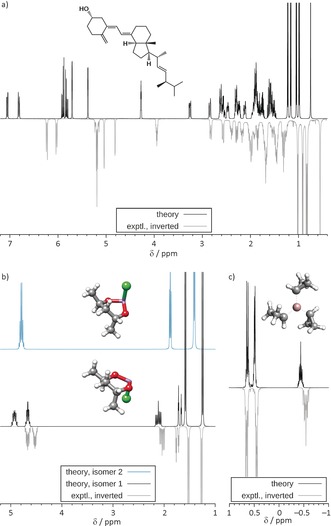
We present a composite procedure for the quantum-chemical computation of spin-spin-coupled H NMR spectra for general, flexible molecules in solution that is based on four main steps, namely conformer/rotamer ensemble (CRE) generation by the fast tight-binding method GFN-xTB and a newly developed search algorithm, computation of the relative free energies and NMR parameters, and solving the spin Hamiltonian. In this way the NMR-specific nuclear permutation problem is solved, and the correct spin symmetries are obtained. Energies, shielding constants, and spin-spin couplings are computed at state-of-the-art DFT levels with continuum solvation. A few (in)organic and transition-metal complexes are presented, and very good, unprecedented agreement between the theoretical and experimental spectra was achieved. The approach is routinely applicable to systems with up to 100-150 atoms and may open new avenues for the detailed (conformational) structure elucidation of, for example, natural products or drug molecules.
我们提出了一种用于计算溶液中一般柔性分子的自旋-自旋耦合 H NMR 谱的组合量子化学计算方法,该方法基于四个主要步骤,即通过快速紧束缚方法 GFN-xTB 和新开发的搜索算法生成构象/旋转异构体(CRE),计算相对自由能和 NMR 参数,并求解自旋哈密顿量。通过这种方式解决了 NMR 特定的核置换问题,并获得了正确的自旋对称性。采用最先进的 DFT 水平和连续溶剂化计算能量、屏蔽常数和自旋-自旋耦合。我们展示了一些(无机)和过渡金属配合物,理论和实验谱之间达到了前所未有的非常好的一致性。该方法通常适用于多达 100-150 个原子的系统,可能为例如天然产物或药物分子的详细(构象)结构阐明开辟新的途径。