Fassio Alexandre V, Martins Pedro M, Guimarães Samuel da S, Junior Sócrates S A, Ribeiro Vagner S, de Melo-Minardi Raquel C, Silveira Sabrina de A
Department of Computer Science, Universidade Federal de Minas Gerais, 6627, Antônio Carlos avenue, Pampulha, Belo Horizonte, 31270-901, Brazil.
Department of Biochemistry and Immunology, Universidade Federal de Minas Gerais, 6627, Antônio Carlos avenue, Pampulha, Belo Horizonte, 31270-901, Brazil.
BMC Bioinformatics. 2017 Sep 13;18(Suppl 10):403. doi: 10.1186/s12859-017-1789-3.
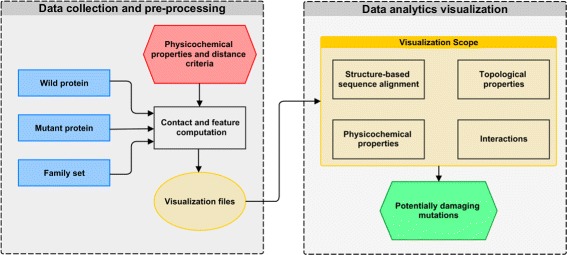
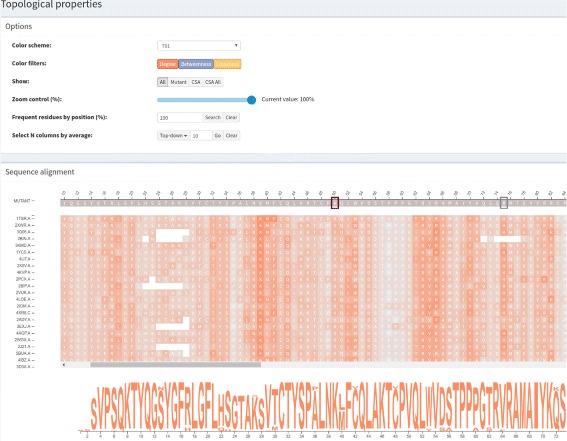
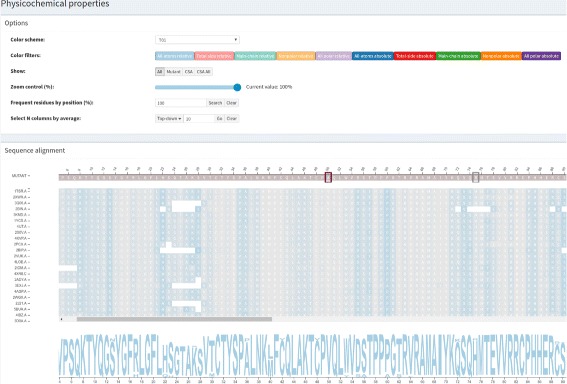
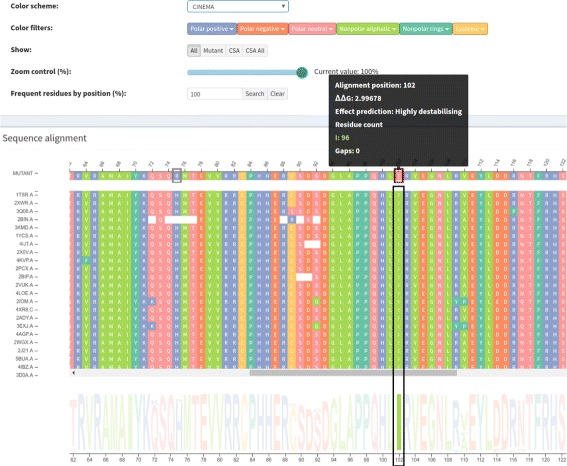
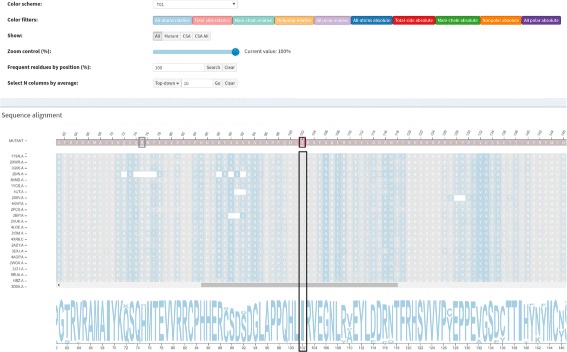
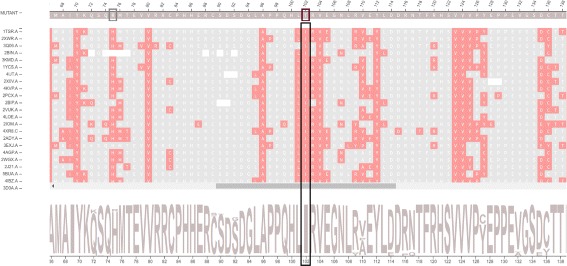
A huge amount of data about genomes and sequence variation is available and continues to grow on a large scale, which makes experimentally characterizing these mutations infeasible regarding disease association and effects on protein structure and function. Therefore, reliable computational approaches are needed to support the understanding of mutations and their impacts. Here, we present VERMONT 2.0, a visual interactive platform that combines sequence and structural parameters with interactive visualizations to make the impact of protein point mutations more understandable.
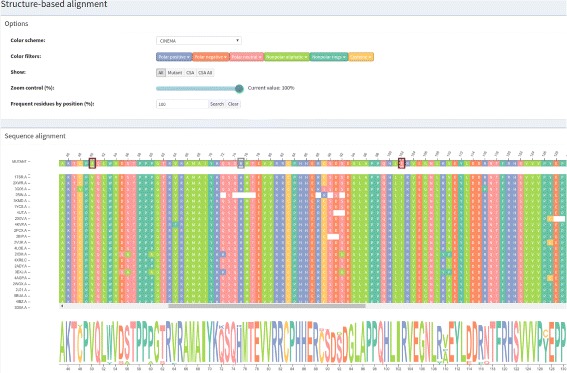
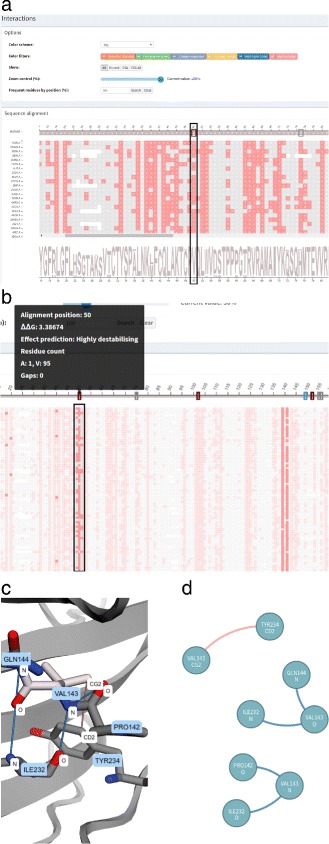
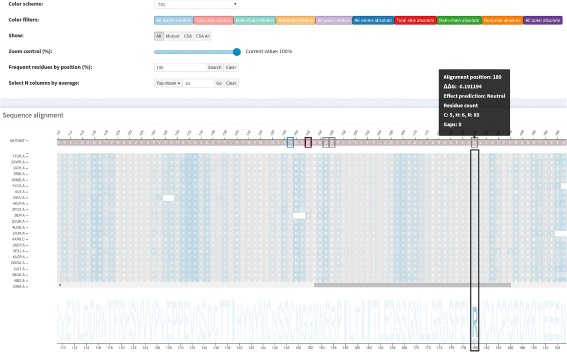
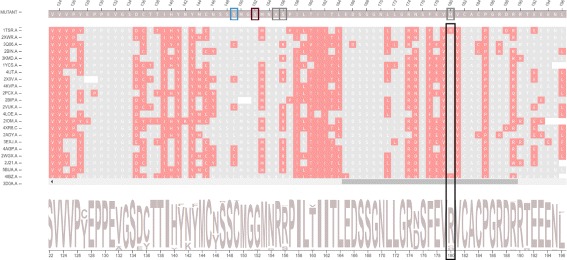
We aimed to contribute a novel visual analytics oriented method to analyze and gain insight on the impact of protein point mutations. To assess the ability of VERMONT to do this, we visually examined a set of mutations that were experimentally characterized to determine if VERMONT could identify damaging mutations and why they can be considered so.
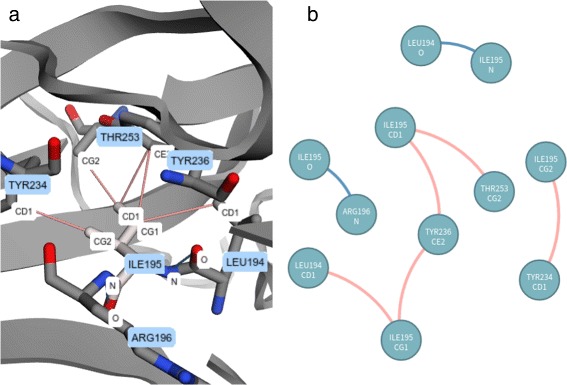
VERMONT allowed us to understand mutations by interpreting position-specific structural and physicochemical properties. Additionally, we note some specific positions we believe have an impact on protein function/structure in the case of mutation.
关于基因组和序列变异的大量数据已经存在,并且仍在大规模持续增长,这使得通过实验来表征这些突变与疾病的关联以及对蛋白质结构和功能的影响变得不可行。因此,需要可靠的计算方法来辅助理解突变及其影响。在此,我们展示了VERMONT 2.0,这是一个视觉交互平台,它将序列和结构参数与交互式可视化相结合,以使蛋白质点突变的影响更易于理解。
我们旨在提供一种新颖的面向视觉分析的方法,以分析和深入了解蛋白质点突变的影响。为了评估VERMONT执行此操作的能力,我们直观地检查了一组经过实验表征的突变,以确定VERMONT是否能够识别有害突变以及为何它们可被视为有害突变。
VERMONT使我们能够通过解释特定位置的结构和物理化学性质来理解突变。此外,我们指出了一些我们认为在发生突变时会对蛋白质功能/结构产生影响的特定位置。