Paulson Joseph N, Chen Cho-Yi, Lopes-Ramos Camila M, Kuijjer Marieke L, Platig John, Sonawane Abhijeet R, Fagny Maud, Glass Kimberly, Quackenbush John
Department of Biostatistics and Computational Biology, Dana-Farber Cancer Institute, Boston, MA, 02215, USA.
Department of Biostatistics, Harvard School of Public Health, Boston, MA, 02215, USA.
BMC Bioinformatics. 2017 Oct 3;18(1):437. doi: 10.1186/s12859-017-1847-x.
Although ultrahigh-throughput RNA-Sequencing has become the dominant technology for genome-wide transcriptional profiling, the vast majority of RNA-Seq studies typically profile only tens of samples, and most analytical pipelines are optimized for these smaller studies. However, projects are generating ever-larger data sets comprising RNA-Seq data from hundreds or thousands of samples, often collected at multiple centers and from diverse tissues. These complex data sets present significant analytical challenges due to batch and tissue effects, but provide the opportunity to revisit the assumptions and methods that we use to preprocess, normalize, and filter RNA-Seq data - critical first steps for any subsequent analysis.
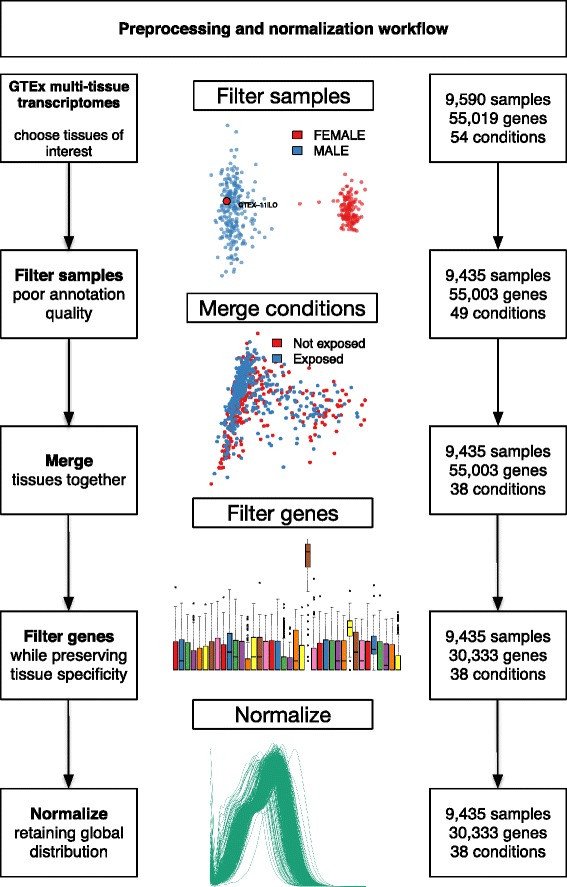
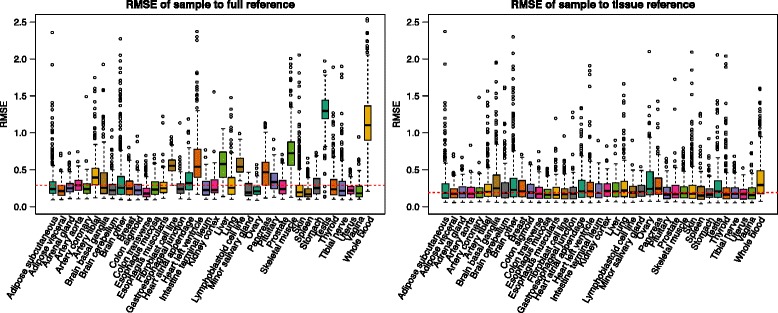
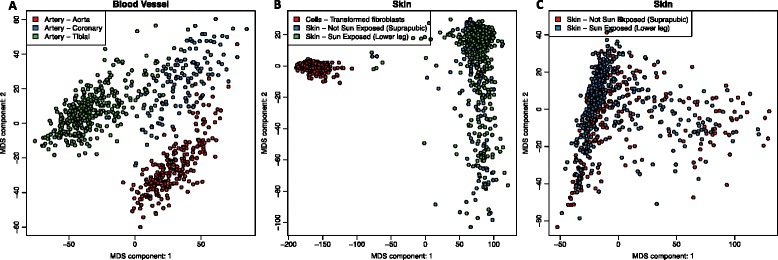
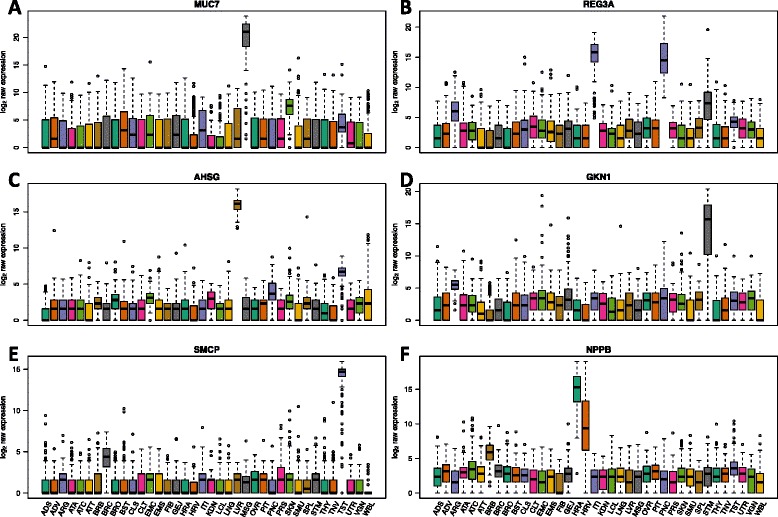
We find that analysis of large RNA-Seq data sets requires both careful quality control and the need to account for sparsity due to the heterogeneity intrinsic in multi-group studies. We developed Yet Another RNA Normalization software pipeline (YARN), that includes quality control and preprocessing, gene filtering, and normalization steps designed to facilitate downstream analysis of large, heterogeneous RNA-Seq data sets and we demonstrate its use with data from the Genotype-Tissue Expression (GTEx) project.
An R package instantiating YARN is available at http://bioconductor.org/packages/yarn .
尽管超高通量RNA测序已成为全基因组转录谱分析的主导技术,但绝大多数RNA测序研究通常仅对数十个样本进行分析,并且大多数分析流程都是针对这些较小规模的研究进行优化的。然而,现在的项目正在生成越来越大的数据集,这些数据集包含来自数百或数千个样本的RNA测序数据,这些样本通常是在多个中心收集的,且来自不同的组织。由于批次和组织效应,这些复杂的数据集带来了重大的分析挑战,但也提供了重新审视我们用于预处理、标准化和过滤RNA测序数据的假设和方法的机会——这是任何后续分析的关键第一步。
我们发现,对大型RNA测序数据集进行分析既需要仔细的质量控制,也需要考虑多组研究中固有的异质性所导致的稀疏性。我们开发了另一种RNA标准化软件流程(YARN),它包括质量控制和预处理、基因过滤以及标准化步骤,旨在促进对大型、异质性RNA测序数据集的下游分析,并且我们展示了其在基因型-组织表达(GTEx)项目数据中的应用。