School of Computer, National University of Defense Technology, Changsha, 410073, China.
Interdiscip Sci. 2022 Mar;14(1):1-14. doi: 10.1007/s12539-021-00473-0. Epub 2021 Sep 6.
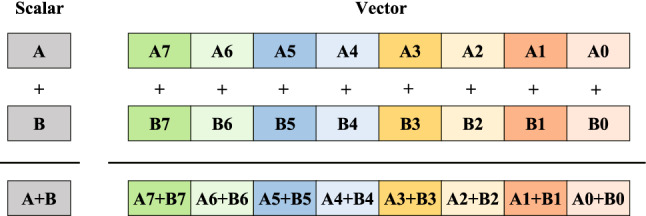
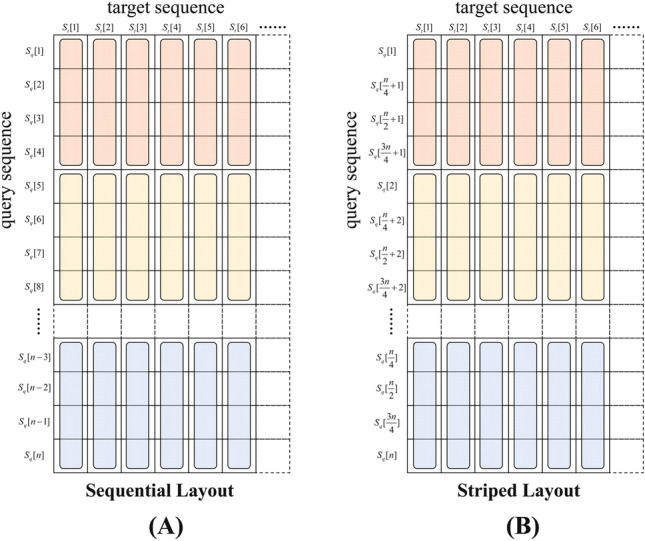
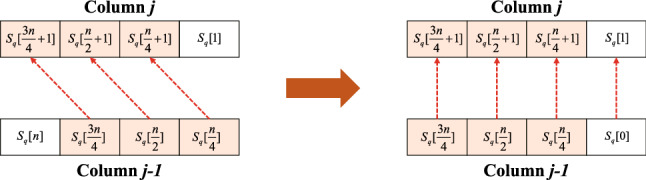
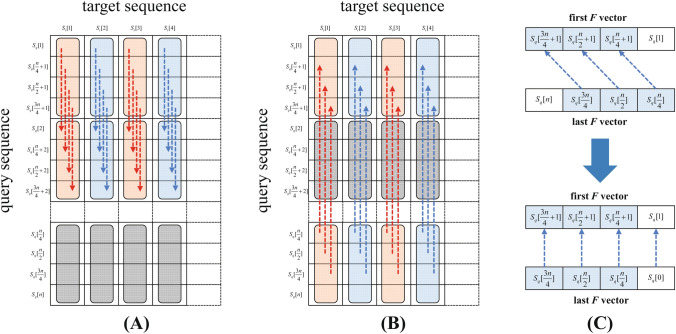
The rapid advances in sequencing technology have led to an explosion of sequence data. Sequence alignment is the central and fundamental problem in many sequence analysis procedure, while local alignment is often the kernel of these algorithms. Usually, Smith-Waterman algorithm is used to find the best subsequence match between given sequences. However, the high time complexity makes the algorithm time-consuming. A lot of approaches have been developed to accelerate and parallelize it, such as vector-level parallelization, thread-level parallelization, process-level parallelization, and heterogeneous acceleration, but the current researches seem unsystematic, which hinders the further research of parallelizing the algorithm. In this paper, we summarize the current research status of parallel local alignments and describe the data layout in these work. Based on the research status, we emphasize large-scale genomic comparisons. By surveying some typical alignment tools' performance, we discuss some possible directions in the future. We hope our work will provide the developers of the alignment tool with technical principle support, and help researchers choose proper alignment tools.
测序技术的快速发展导致了序列数据的爆炸式增长。序列比对是许多序列分析过程中的核心和基本问题,而局部比对通常是这些算法的核心。通常,Smith-Waterman 算法用于在给定序列之间找到最佳子序列匹配。然而,高时间复杂度使得算法非常耗时。已经开发了许多方法来加速和并行化它,例如矢量级并行化、线程级并行化、进程级并行化和异构加速,但目前的研究似乎缺乏系统性,这阻碍了算法的进一步研究。在本文中,我们总结了并行局部比对的当前研究现状,并描述了这些工作中的数据布局。基于研究现状,我们强调了大规模基因组比较。通过调查一些典型比对工具的性能,我们讨论了未来的一些可能方向。我们希望我们的工作将为比对工具的开发人员提供技术原理支持,并帮助研究人员选择合适的比对工具。