Earl Rachel K, Turner Tychele N, Mefford Heather C, Hudac Caitlin M, Gerdts Jennifer, Eichler Evan E, Bernier Raphael A
Department of Psychiatry and Behavioral Sciences, University of Washington, CHDD Box 357920, Seattle, WA 98195 USA.
Department of Genome Sciences, University of Washington, Seattle, WA USA.
Mol Autism. 2017 Oct 5;8:54. doi: 10.1186/s13229-017-0173-5. eCollection 2017.
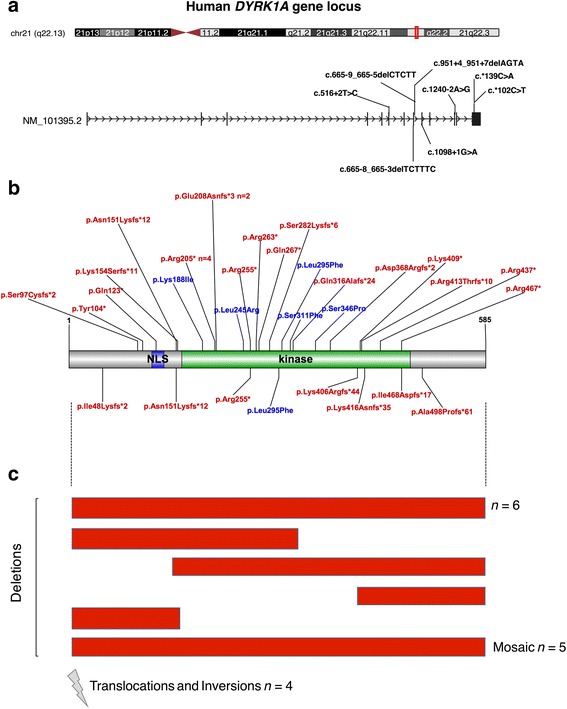
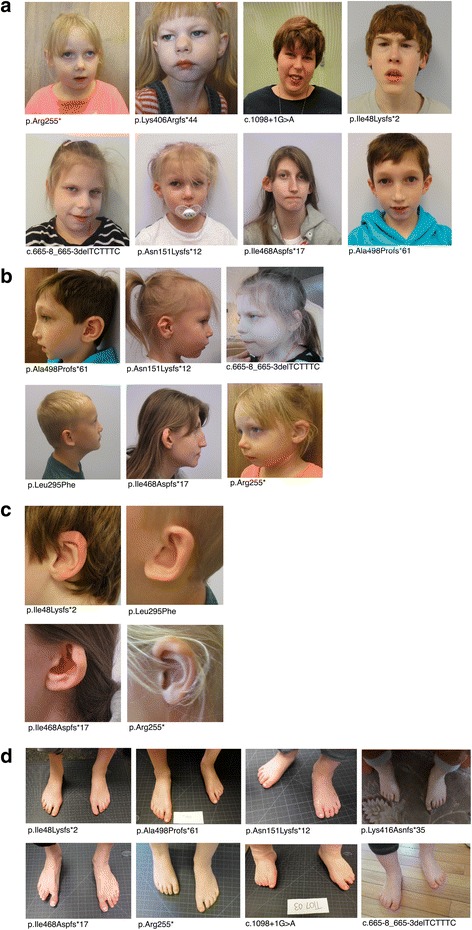
is a gene recurrently disrupted in 0.1-0.5% of the ASD population. A growing number of case reports with haploinsufficiency exhibit common phenotypic features including microcephaly, intellectual disability, speech delay, and facial dysmorphisms.
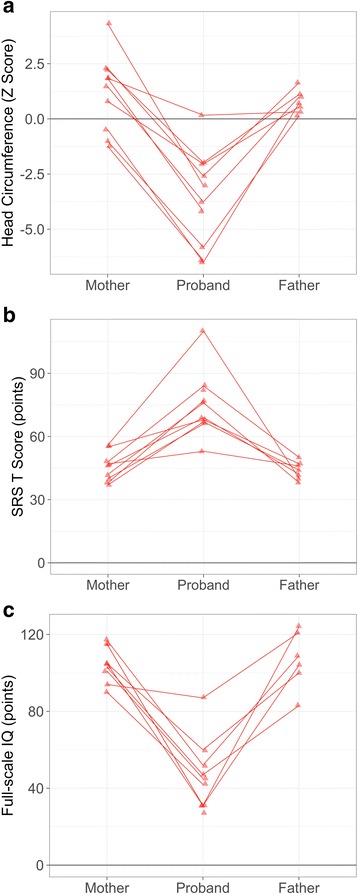
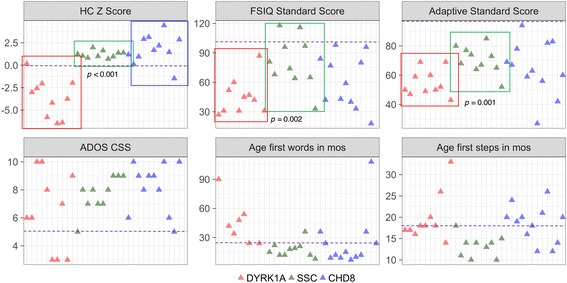
Phenotypic information from previously published cases ( = 51) and participants in an ongoing study at the University of Washington (UW, = 10) were compiled. Frequencies of recurrent phenotypic features in this population were compared to features observed in a large sample with idiopathic ASD from the Simons Simplex Collection ( = 1981). UW cases were further characterized quantitatively and compared to a randomly subsampled set of idiopathic ASD cases matched on age and gender ( = 10) and to cases with an ASD-associated disruptive mutation to ( = 12). Contribution of familial genetic background to clinical heterogeneity was assessed by comparing head circumference, IQ, and ASD-related symptoms of UW cases to their unaffected parents.
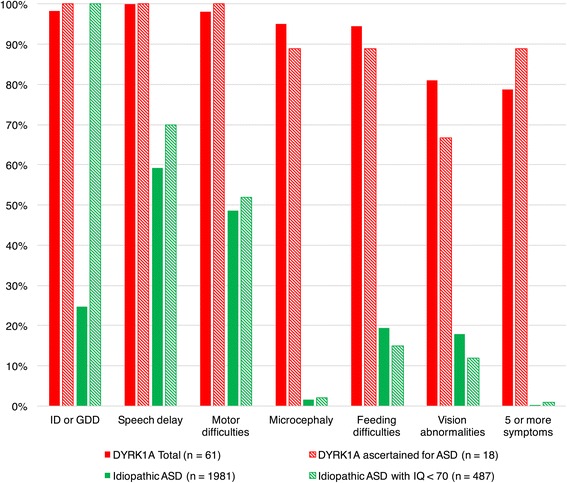
haploinsufficiency results in a common phenotypic profile including intellectual disability, speech and motor difficulties, microcephaly, feeding difficulties, and vision abnormalities. Eighty-nine percent of cases ascertained for ASD presented with a constellation of five or more of these symptoms. When compared quantitatively, cases presented with significantly lower IQ and adaptive functioning compared to idiopathic cases and significantly smaller head size compared to both idiopathic and cases. Phenotypic variability in parental head circumference, IQ, and ASD-related symptoms corresponded to observed variability in affected child phenotype.
Results confirm a core clinical phenotype for disruptions, with a combination of features that is distinct from idiopathic ASD. Cases with mutations are also distinguishable from disruptive mutations to by head size. Measurable, quantitative characterization of haploinsufficiency illuminates clinical variability, which may be, in part, due to familial genetic background.
某基因在0.1% - 0.5%的自闭症谱系障碍(ASD)人群中反复出现破坏情况。越来越多关于该基因单倍剂量不足的病例报告显示出常见的表型特征,包括小头畸形、智力残疾、语言发育迟缓以及面部畸形。
收集了先前发表的病例(n = 51)以及华盛顿大学(UW)正在进行的一项研究中的参与者(n = 10)的表型信息。将该人群中反复出现的表型特征频率与来自西蒙斯单纯型病例集的大量特发性ASD样本(n = 1981)中观察到的特征进行比较。对UW病例进行了进一步的定量特征分析,并与一组按年龄和性别匹配的随机抽取的特发性ASD病例(n = 10)以及携带与ASD相关的该基因破坏突变的病例(n = 12)进行比较。通过比较UW病例与其未受影响的父母的头围、智商和与ASD相关的症状,评估家族遗传背景对临床异质性的贡献。
该基因单倍剂量不足导致了一种常见表型特征,包括智力残疾、语言和运动困难、小头畸形、喂养困难以及视力异常。被确定为患有ASD的该基因相关病例中,89%表现出五种或更多这些症状。在进行定量比较时,与特发性病例相比,该基因相关病例的智商和适应性功能显著更低,与特发性病例和该基因其他突变病例相比,头围显著更小。父母的头围、智商和与ASD相关症状的表型变异性与受影响儿童表型中观察到的变异性相对应。
结果证实了该基因破坏的核心临床表型,其特征组合与特发性ASD不同。该基因突变病例在头围方面也与该基因其他破坏突变病例有所区别。对该基因单倍剂量不足进行可测量的定量特征分析揭示了临床变异性,这可能部分归因于家族遗传背景。