Section of Stress, Psychiatry and Immunology and Perinatal Psychiatry, King's College London, London, United Kingdom.
Institute of Psychiatry, Psychology and Neuroscience, Department of Psychological Medicine, London, United Kingdom.
Int J Neuropsychopharmacol. 2018 Feb 1;21(2):187-200. doi: 10.1093/ijnp/pyx083.
In humans, interferon-α treatment for chronic viral hepatitis is a well-recognized clinical model for inflammation-induced depression, but the molecular mechanisms underlying these effects are not clear. Following peripheral administration in rodents, interferon-α induces signal transducer and activator of transcription-1 (STAT1) within the hippocampus and disrupts hippocampal neurogenesis.
We used the human hippocampal progenitor cell line HPC0A07/03C to evaluate the effects of 2 concentrations of interferon-α, similar to those observed in human serum during its therapeutic use (500 pg/mL and 5000 pg/mL), on neurogenesis and apoptosis.
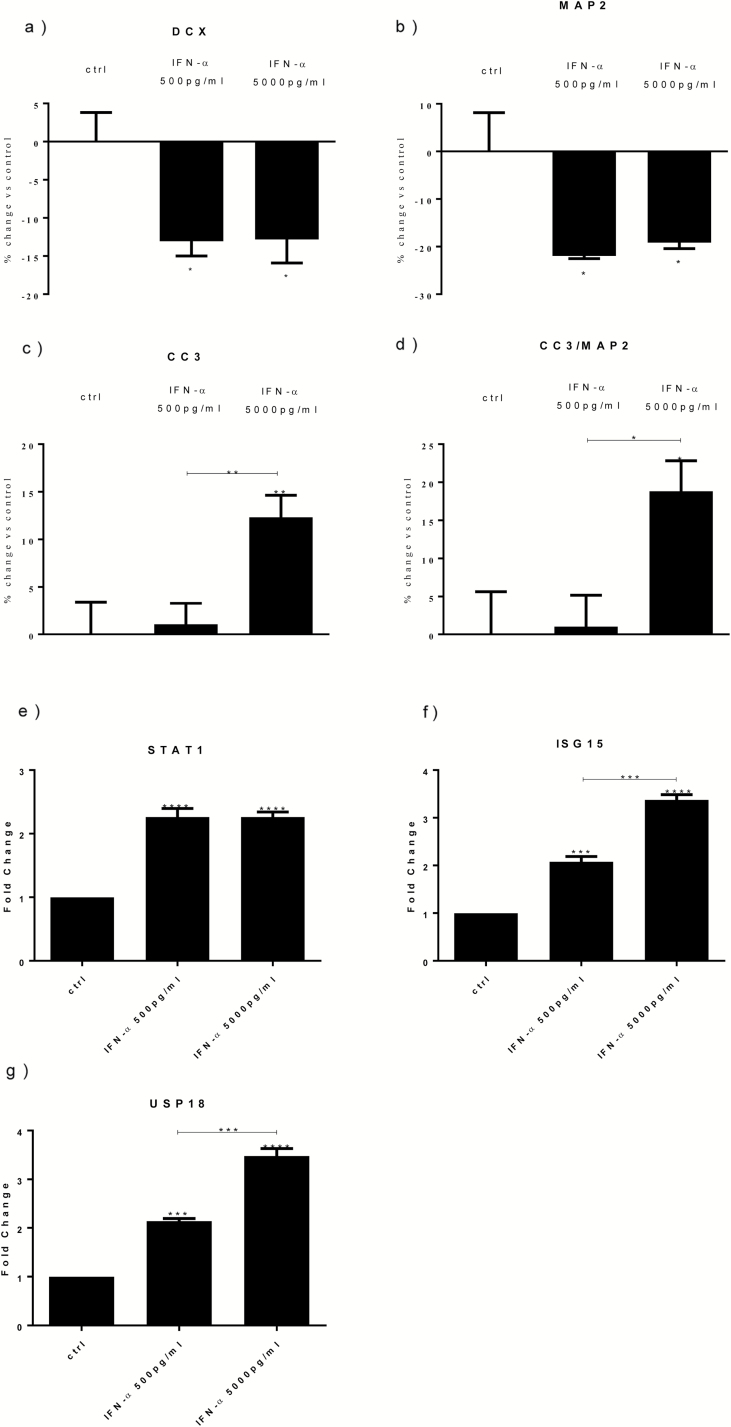
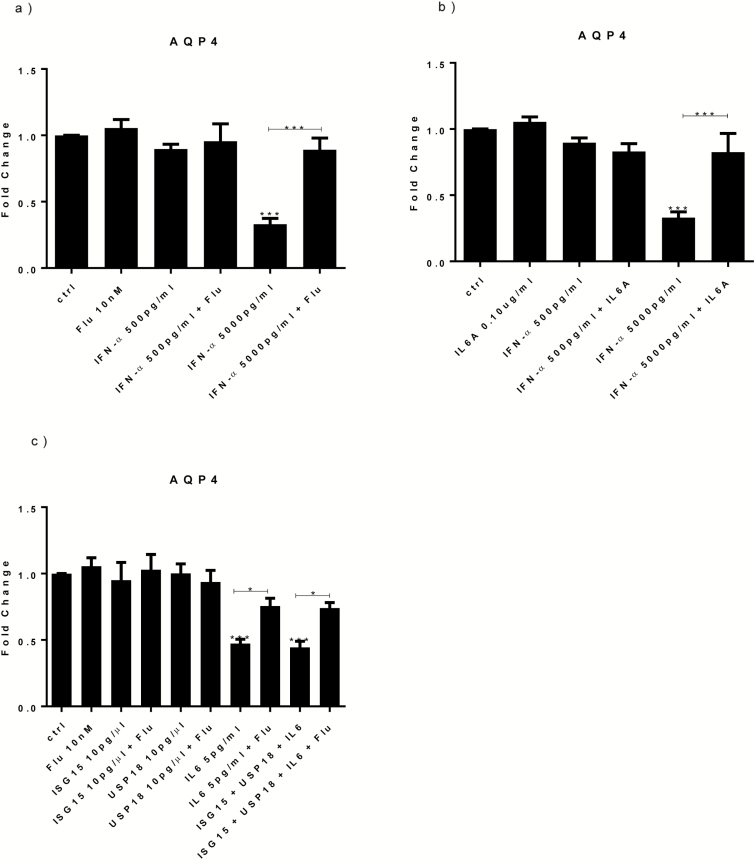
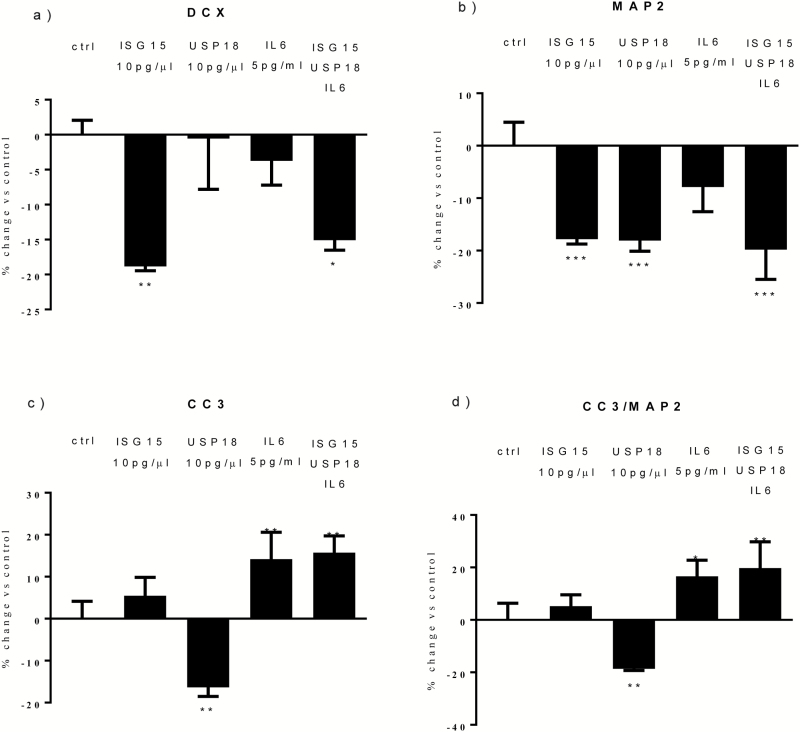
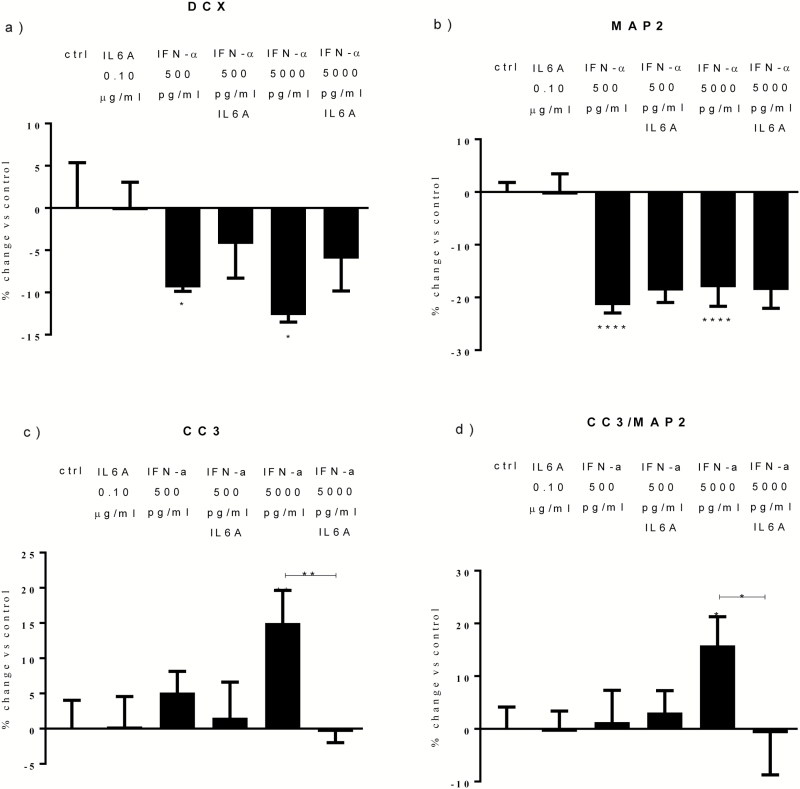
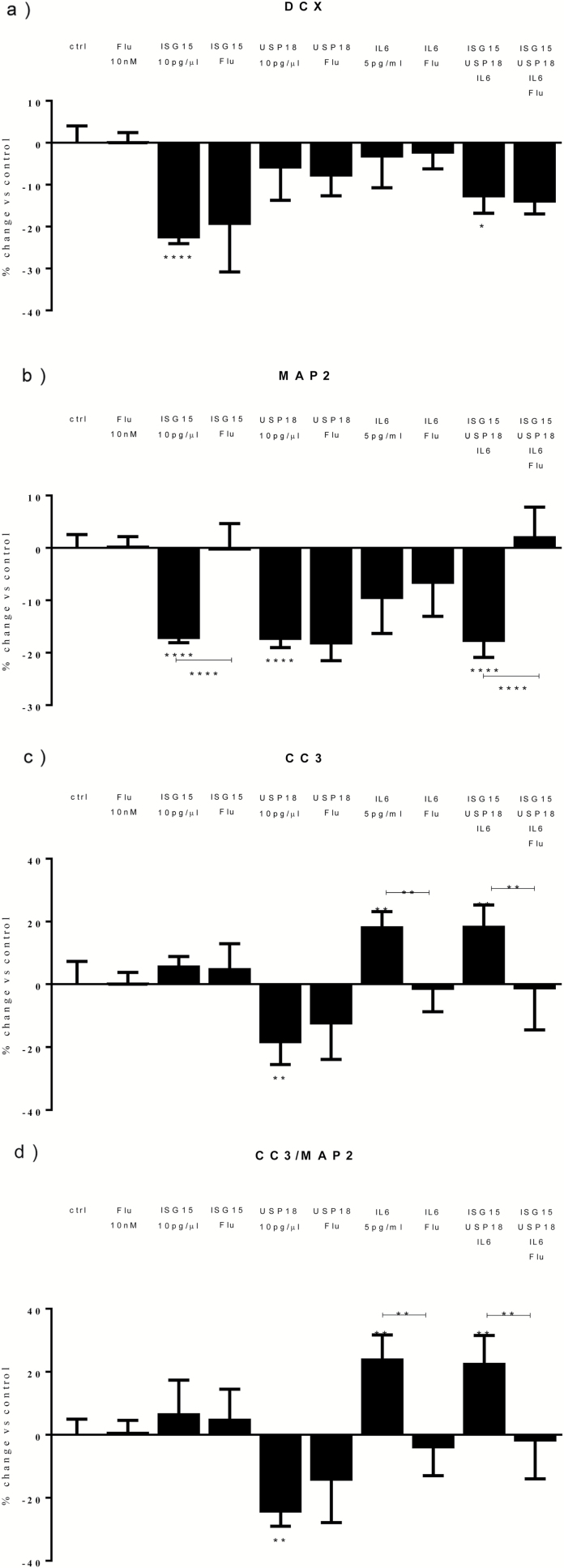
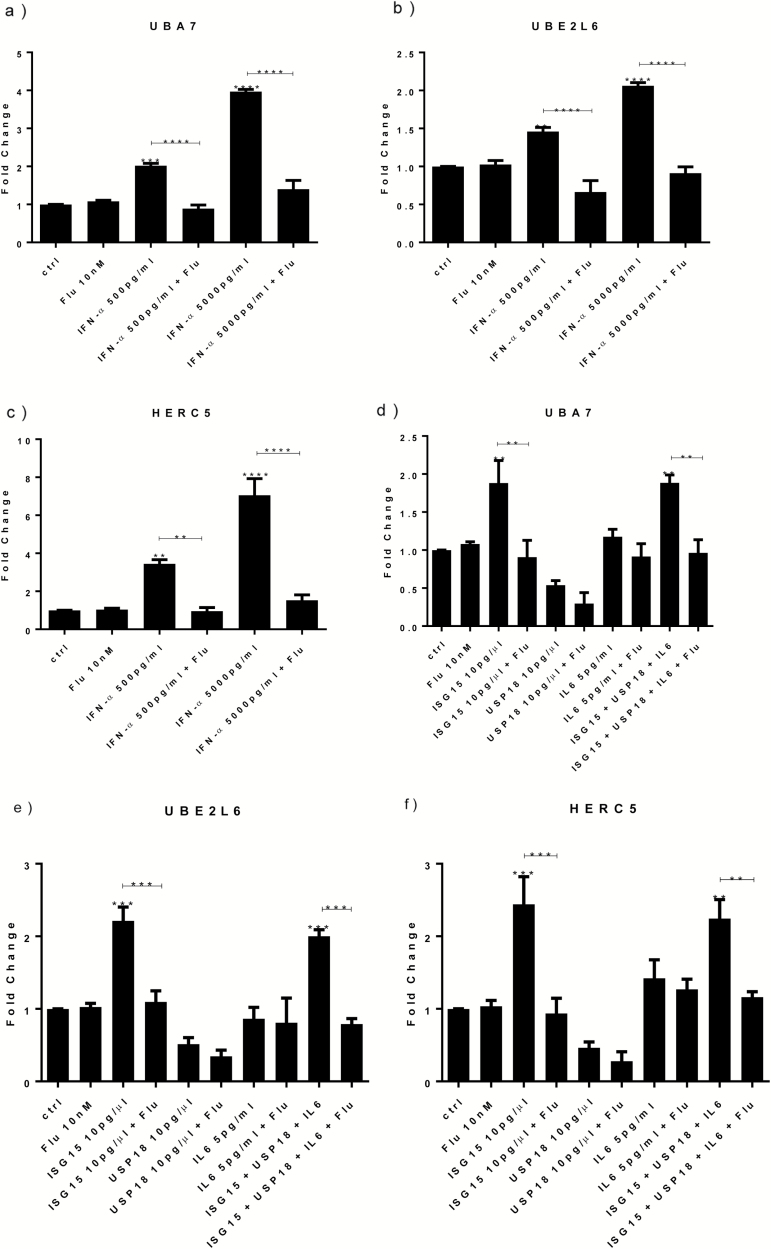
Both concentrations of interferon-α decreased hippocampal neurogenesis, with the high concentration also increasing apoptosis. Moreover, interferon-α increased the expression of interferon-stimulated gene 15 (ISG15), ubiquitin-specific peptidase 18 (USP18), and interleukin-6 (IL-6) via activation of STAT1. Like interferon-α, co-treatment with a combination of ISG15, USP18, and IL-6 was able to reduce neurogenesis and enhance apoptosis via further downstream activation of STAT1. Further experiments showed that ISG15 and USP18 mediated the interferon-α-induced reduction in neurogenesis (potentially through upregulation of the ISGylation-related proteins UBA7, UBE2L6, and HERC5), while IL-6 mediated the interferon-α-induced increase in apoptosis (potentially through downregulation of aquaporin 4). Using transcriptomic analyses, we showed that interferon-α regulated pathways involved in oxidative stress and immune response (e.g., Nuclear Factor (erythroid-derived 2)-like 2 [Nrf2] and interferon regulatory factor [IRF] signaling pathway), neuronal formation (e.g., CAMP response element-binding protein [CREB] signaling), and cell death regulation (e.g., tumor protein(p)53 signaling).
We identify novel molecular mechanisms mediating the effects of interferon-α on the human hippocampus potentially involved in inflammation-induced neuropsychiatric symptoms.
在人类中,干扰素-α治疗慢性病毒性肝炎是一种公认的炎症诱导抑郁的临床模型,但这些影响的分子机制尚不清楚。在啮齿动物外周给药后,干扰素-α在海马体内诱导信号转导和转录激活因子 1(STAT1),并破坏海马神经发生。
我们使用人海马祖细胞系 HPC0A07/03C 来评估两种浓度的干扰素-α(类似于其在治疗用途中在人血清中观察到的浓度(500pg/mL 和 5000pg/mL))对神经发生和细胞凋亡的影响。
两种浓度的干扰素-α均降低了海马神经发生,高浓度也增加了细胞凋亡。此外,干扰素-α通过激活 STAT1,增加干扰素刺激基因 15(ISG15)、泛素特异性肽酶 18(USP18)和白细胞介素 6(IL-6)的表达。与干扰素-α一样,ISG15、USP18 和 IL-6 的联合治疗也能够通过进一步下游激活 STAT1 来减少神经发生并增强细胞凋亡。进一步的实验表明,ISG15 和 USP18 介导了干扰素-α诱导的神经发生减少(可能通过上调 ISGylation 相关蛋白 UBA7、UBE2L6 和 HERC5),而 IL-6 介导了干扰素-α诱导的细胞凋亡增加(可能通过下调水通道蛋白 4)。通过转录组分析,我们表明干扰素-α调节了涉及氧化应激和免疫反应的途径(例如,核因子(红细胞衍生 2)样 2(Nrf2)和干扰素调节因子(IRF)信号通路)、神经元形成(例如,cAMP 反应元件结合蛋白(CREB)信号通路)和细胞死亡调节(例如,肿瘤蛋白(p)53 信号通路)。
我们确定了介导干扰素-α对人海马体影响的新分子机制,这些机制可能与炎症诱导的神经精神症状有关。