Department of Statistical Science, Southern Methodist University, Dallas, TX 75275, USA.
Quantitative Biomedical Research Center, Center for the Genetics of Host Defense, Department of Clinical Science, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Stat Med. 2018 Feb 20;37(4):659-672. doi: 10.1002/sim.7540. Epub 2017 Oct 19.
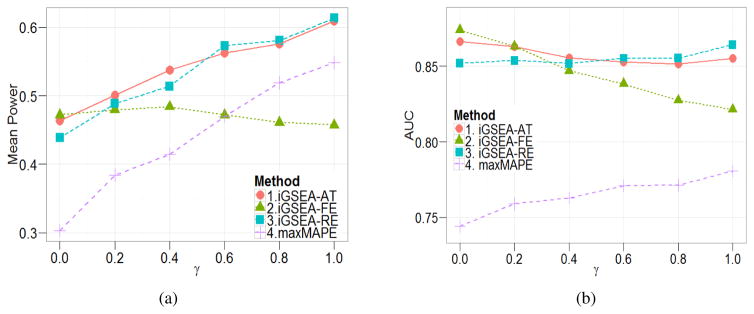
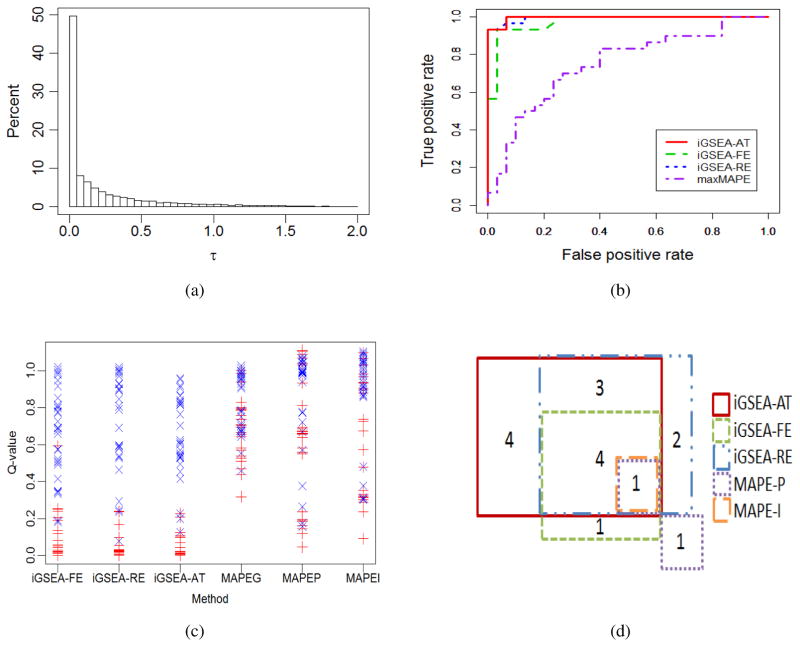
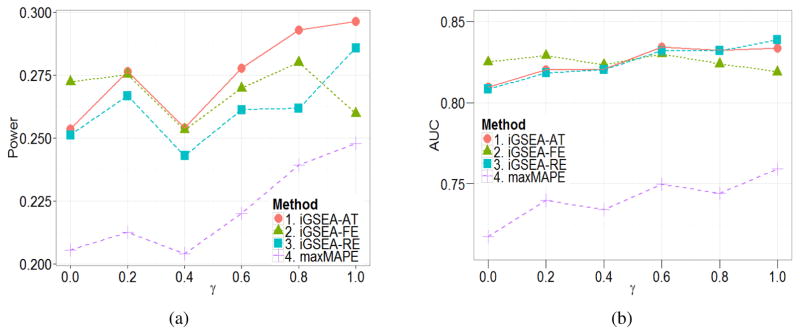
In the field of gene set enrichment analysis (GSEA), meta-analysis has been used to integrate information from multiple studies to present a reliable summarization of the expanding volume of individual biomedical research, as well as improve the power of detecting essential gene sets involved in complex human diseases. However, existing methods, Meta-Analysis for Pathway Enrichment (MAPE), may be subject to power loss because of (1) using gross summary statistics for combining end results from component studies and (2) using enrichment scores whose distributions depend on the set sizes. In this paper, we adapt meta-analysis approaches recently developed for genome-wide association studies, which are based on fixed effect and random effects (RE) models, to integrate multiple GSEA studies. We further develop a mixed strategy via adaptive testing for choosing RE versus FE models to achieve greater statistical efficiency as well as flexibility. In addition, a size-adjusted enrichment score based on a one-sided Kolmogorov-Smirnov statistic is proposed to formally account for varying set sizes when testing multiple gene sets. Our methods tend to have much better performance than the MAPE methods and can be applied to both discrete and continuous phenotypes. Specifically, the performance of the adaptive testing method seems to be the most stable in general situations.
在基因集富集分析(GSEA)领域,荟萃分析已被用于整合来自多个研究的信息,以可靠地总结不断增加的个体生物医学研究,并提高检测复杂人类疾病中涉及的重要基因集的能力。然而,现有的方法 Meta-Analysis for Pathway Enrichment(MAPE)可能会由于以下原因而失去功效:(1)使用总体汇总统计信息来组合来自组成研究的最终结果;(2)使用依赖于集合大小的富集分数。在本文中,我们将最近为全基因组关联研究开发的荟萃分析方法进行了改编,这些方法基于固定效应和随机效应(RE)模型,以整合多个 GSEA 研究。我们进一步通过自适应检验开发了一种混合策略,用于选择 RE 与 FE 模型,以实现更高的统计效率和灵活性。此外,还提出了一种基于单边柯尔莫哥洛夫-斯米尔诺夫统计量的大小调整后的富集分数,以在测试多个基因集时正式考虑集合大小的变化。我们的方法往往比 MAPE 方法具有更好的性能,并且可应用于离散和连续表型。具体而言,自适应检验方法的性能在一般情况下似乎最为稳定。