Centre de recherche du Centre hospitalier de l'Université de Montréal (CRCHUM) Montréal, Québec H2X 0A9, Canada.
Département de biochimie et médecine moléculaire, Université de Montréal, Montréal, Québec H3T 1J4, Canada.
Dis Model Mech. 2017 Dec 19;10(12):1465-1480. doi: 10.1242/dmm.029736.
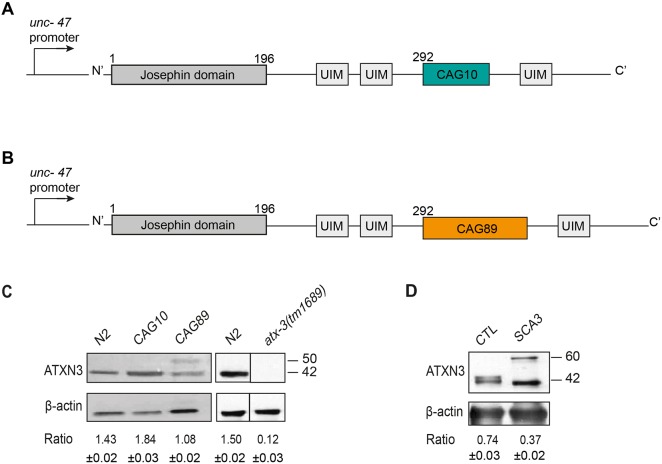
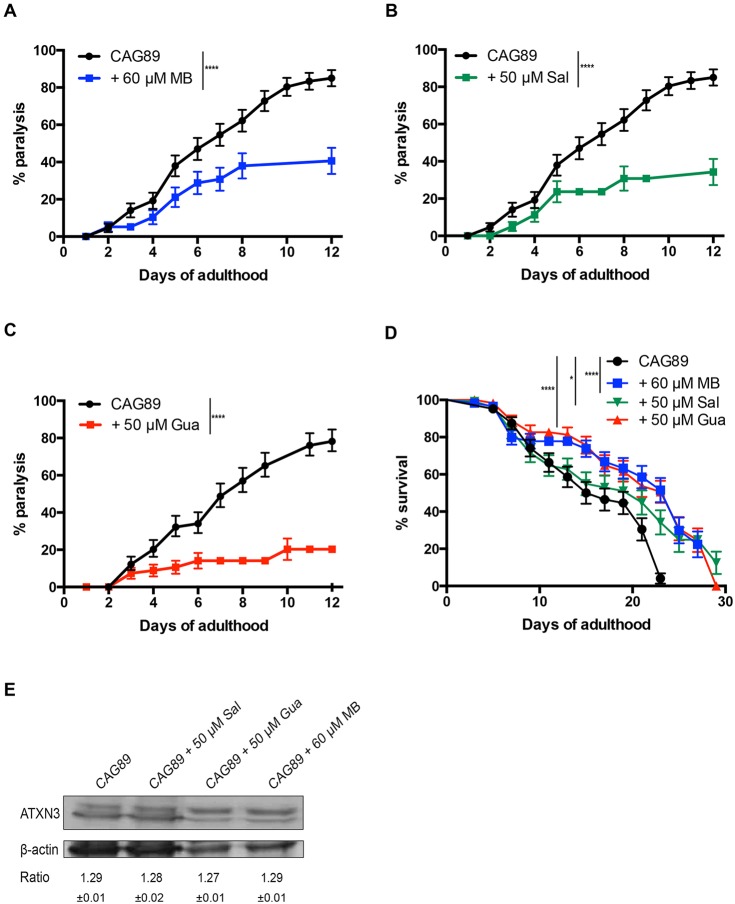
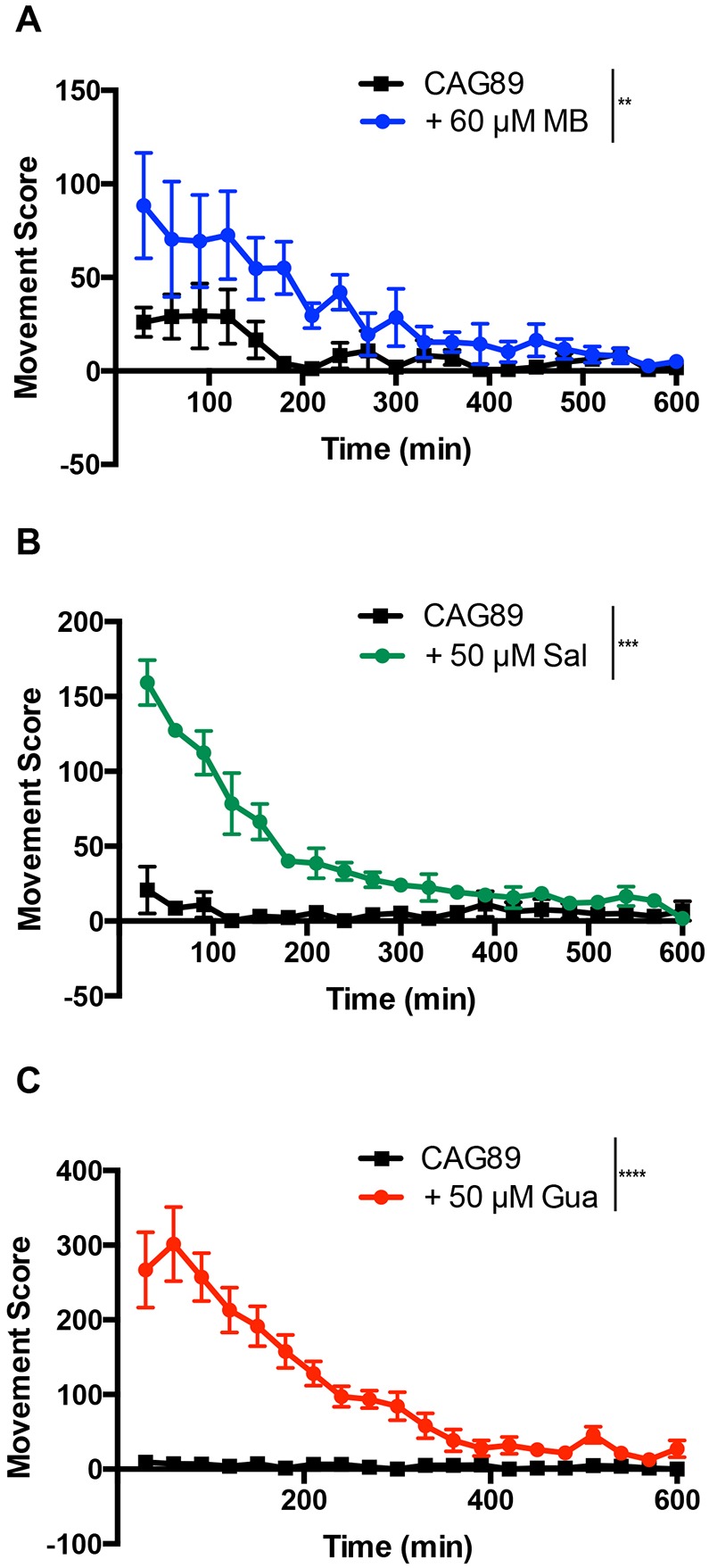
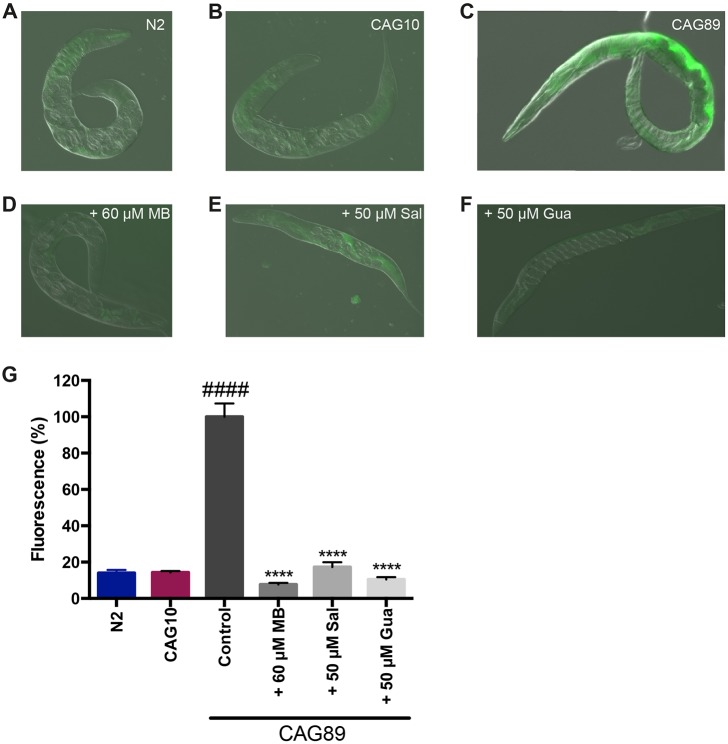
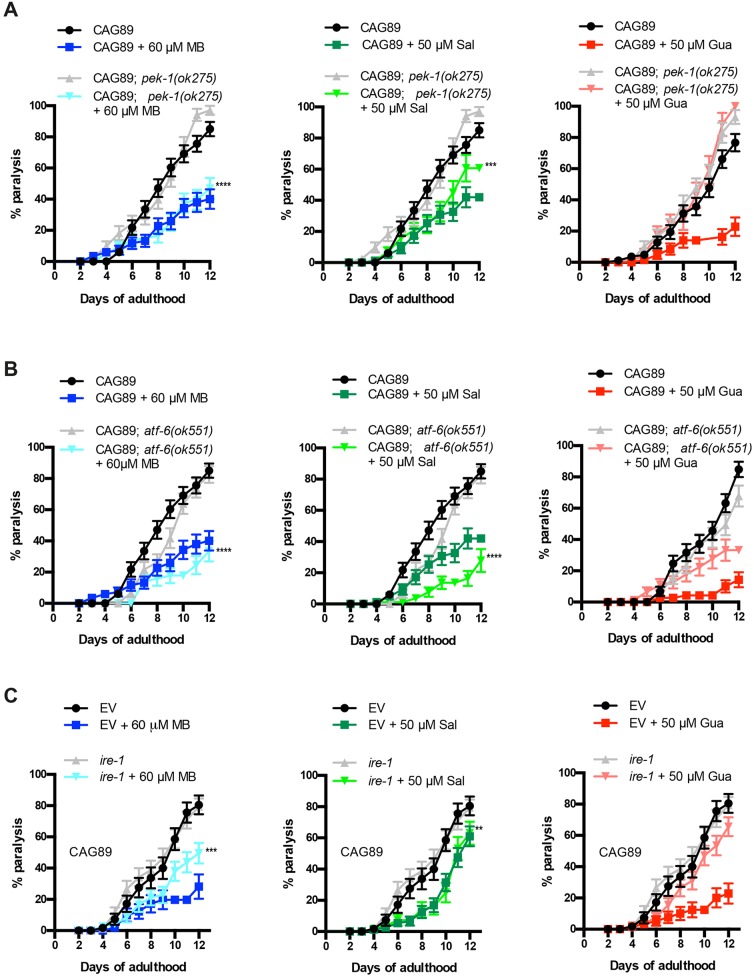
Polyglutamine expansion diseases are a group of hereditary neurodegenerative disorders that develop when a CAG repeat in the causative genes is unstably expanded above a certain threshold. The expansion of trinucleotide CAG repeats causes hereditary adult-onset neurodegenerative disorders, such as Huntington's disease, dentatorubral-pallidoluysian atrophy, spinobulbar muscular atrophy and multiple forms of spinocerebellar ataxia (SCA). The most common dominantly inherited SCA is the type 3 (SCA3), also known as Machado-Joseph disease (MJD), which is an autosomal dominant, progressive neurological disorder. The gene causatively associated with MJD is Recent studies have shown that this gene modulates endoplasmic reticulum (ER) stress. We generated transgenic strains expressing human genes in motoneurons, and animals expressing mutant alleles showed decreased lifespan, impaired movement, and rates of neurodegeneration greater than wild-type controls. We tested three neuroprotective compounds (Methylene Blue, guanabenz and salubrinal) believed to modulate ER stress and observed that these molecules rescued phenotypes. Furthermore, these compounds required specific branches of the ER unfolded protein response (UPR), reduced global ER and oxidative stress, and polyglutamine aggregation. We introduce new models for MJD based on the expression of full-length in a limited number of neurons. Using these models, we discovered that chemical modulation of the UPR reduced neurodegeneration and warrants investigation in mammalian models of MJD.
多聚谷氨酰胺扩展疾病是一组遗传性神经退行性疾病,当致病基因中的 CAG 重复不稳定扩展超过一定阈值时就会发生。三核苷酸 CAG 重复扩展引起遗传性成人发病的神经退行性疾病,如亨廷顿病、齿状核红核苍白球路易体萎缩、延髓脊髓性肌萎缩和多种脊髓小脑共济失调(SCA)。最常见的显性遗传 SCA 是 3 型(SCA3),也称为 Machado-Joseph 病(MJD),是一种常染色体显性、进行性神经系统疾病。与 MJD 相关的基因是 最近的研究表明,该基因调节内质网(ER)应激。我们生成了在运动神经元中表达人类 基因的转基因 株系,表达突变 等位基因的动物表现出寿命缩短、运动障碍和神经退行性变速度高于野生型 对照。我们测试了三种被认为能调节 ER 应激的神经保护化合物(亚甲蓝、胍那苄和 salubrinal),并观察到这些分子挽救了 表型。此外,这些化合物需要特定的内质网未折叠蛋白反应(UPR)分支,减少全局 ER 和氧化应激,并减少多聚谷氨酰胺聚集。我们引入了新的 MJD 模型,基于全长 在有限数量的神经元中的表达。使用这些模型,我们发现 UPR 的化学调节减少了神经退行性变,并值得在 MJD 的哺乳动物模型中进行研究。