Gao Rui, Liu Yongping, Silva-Fernandes Anabela, Fang Xiang, Paulucci-Holthauzen Adriana, Chatterjee Arpita, Zhang Hang L, Matsuura Tohru, Choudhary Sanjeev, Ashizawa Tetsuo, Koeppen Arnulf H, Maciel Patricia, Hazra Tapas K, Sarkar Partha S
Department of Neurology, University of Texas Medical Branch, Galveston, Texas, United States of America.
Life and Health Sciences Research Institute (ICVS), School of Health Sciences, University of Minho, Braga, Portugal; ICVS/3B's PT Government Associate Laboratory, Braga/Guimarặes, Portugal.
PLoS Genet. 2015 Jan 15;11(1):e1004834. doi: 10.1371/journal.pgen.1004834. eCollection 2015 Jan.
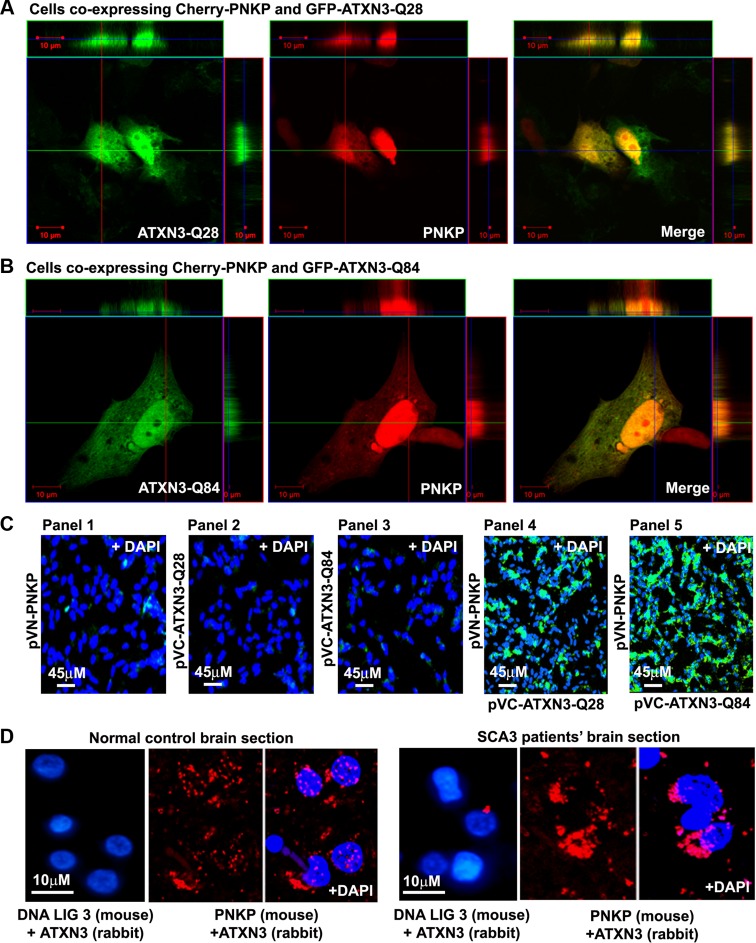
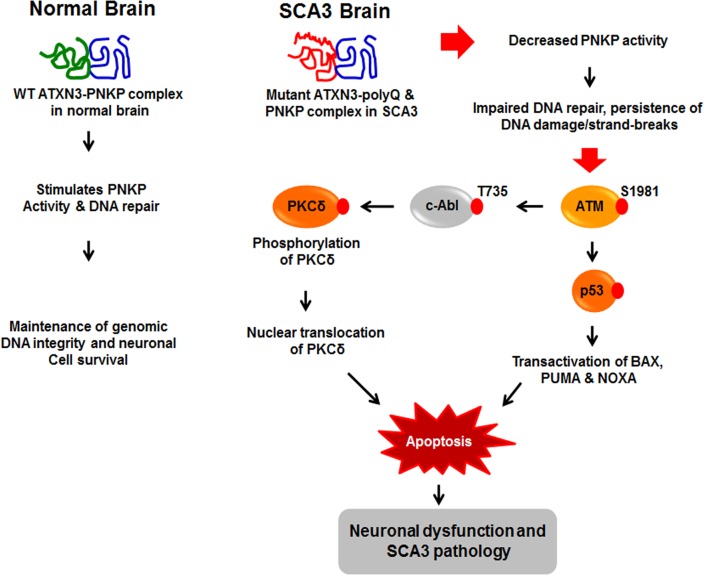
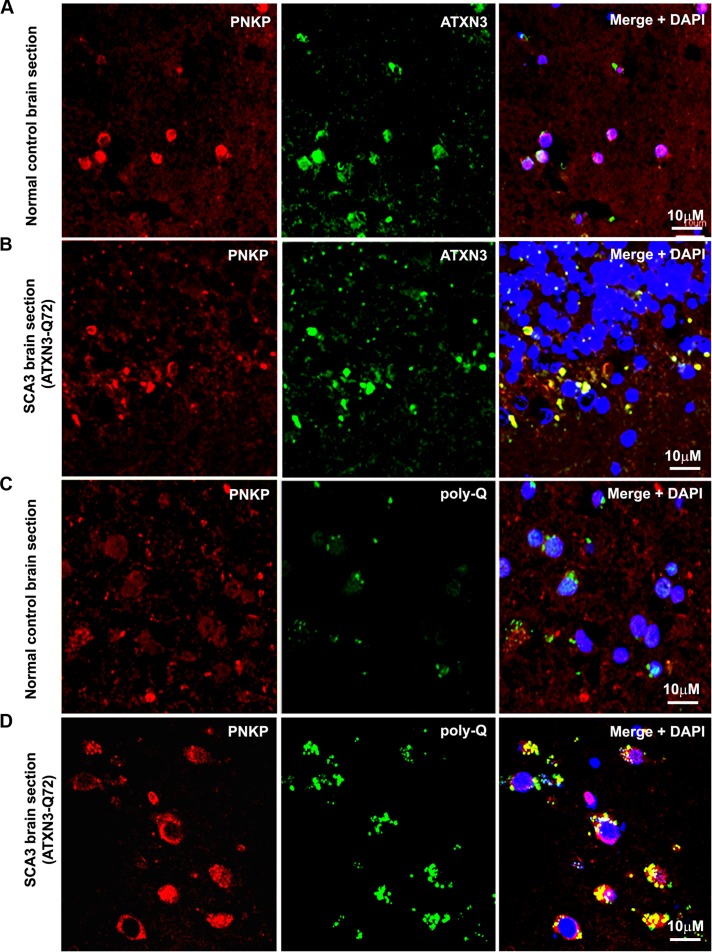
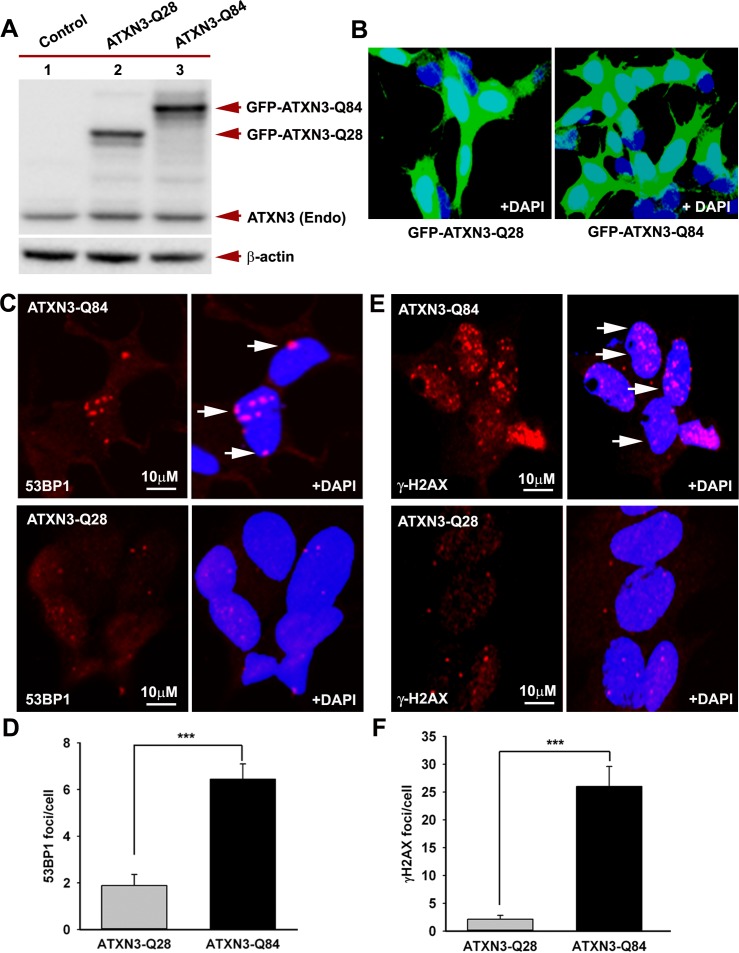
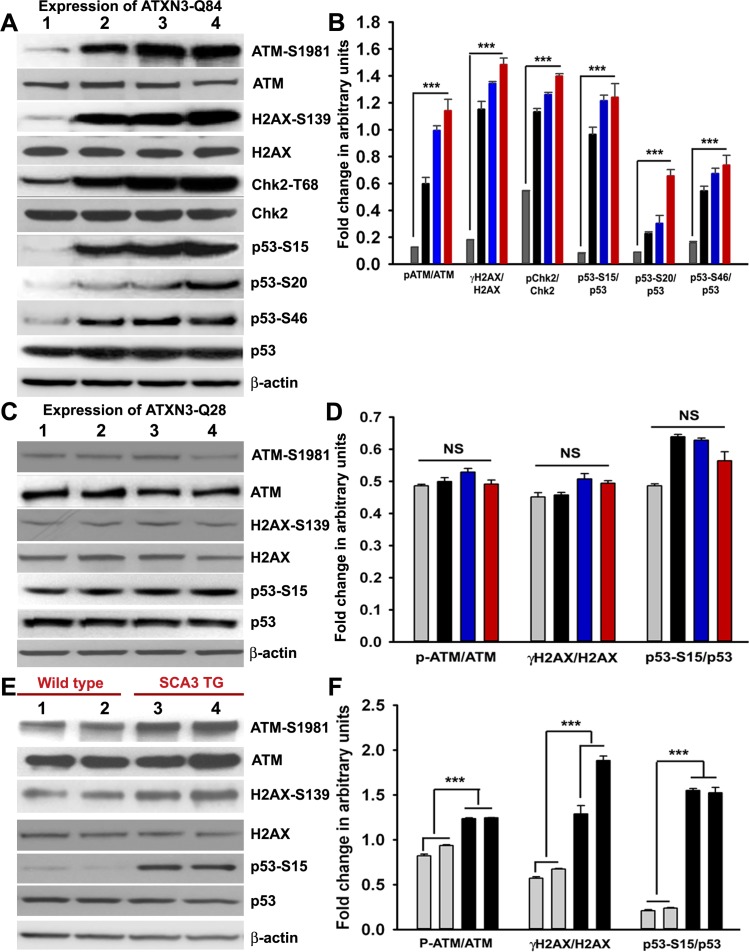
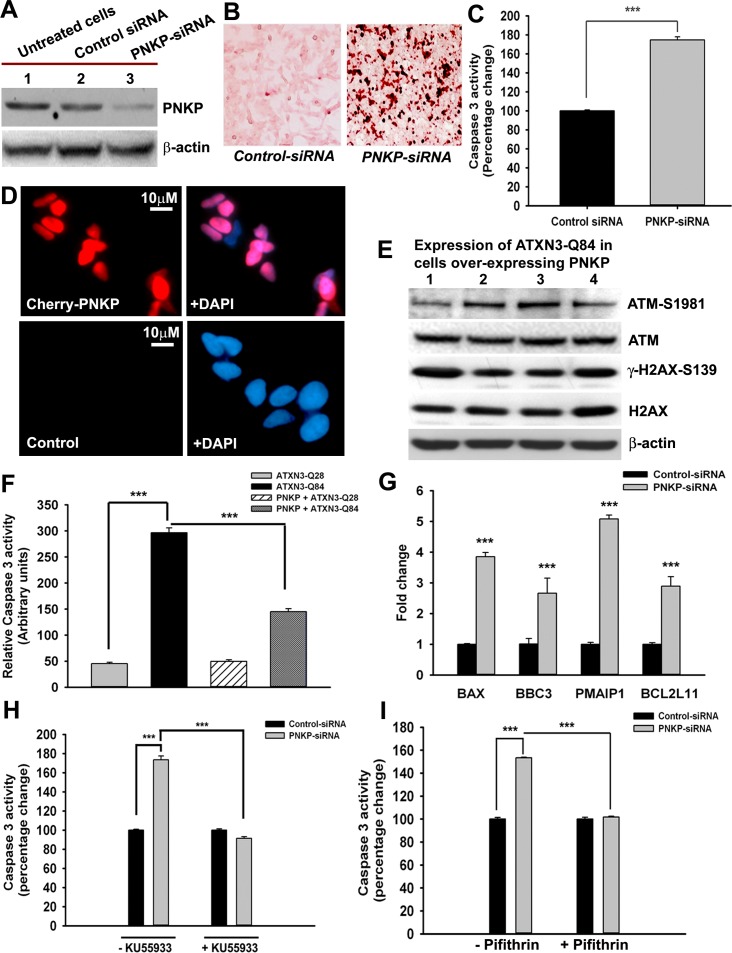
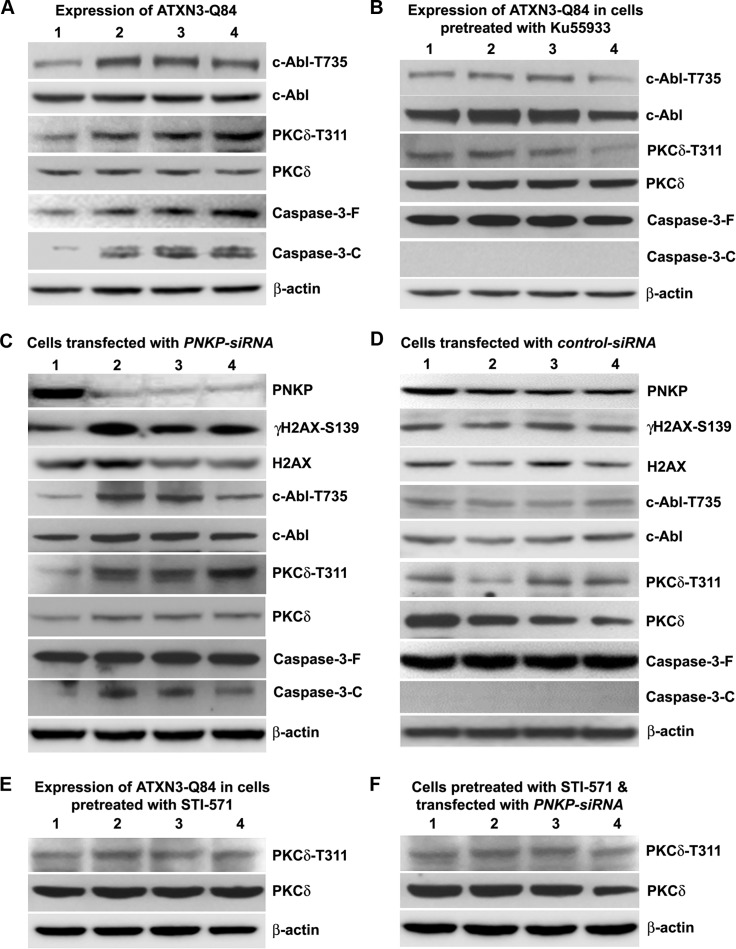
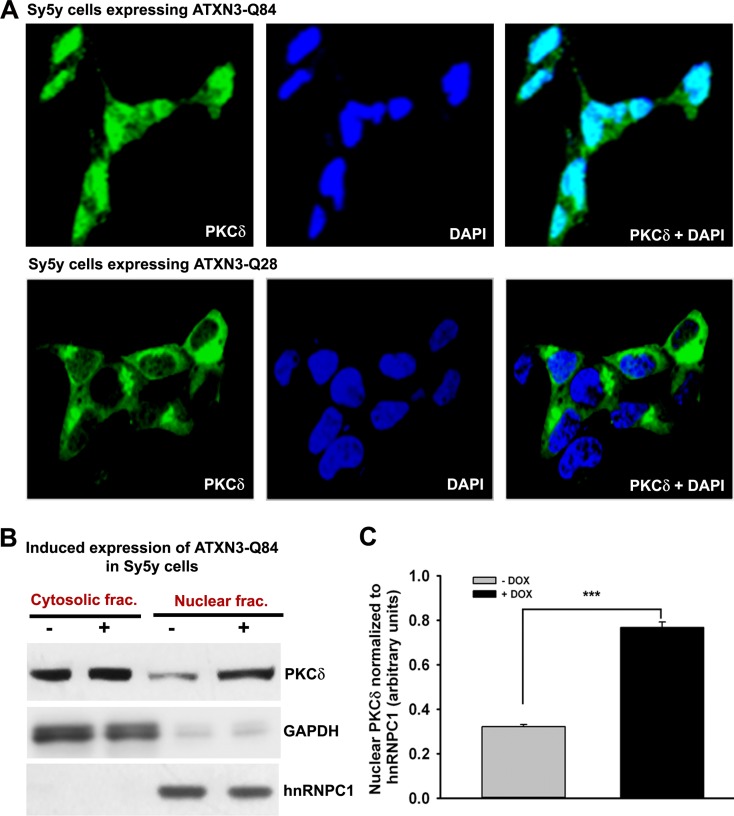
Spinocerebellar ataxia type 3 (SCA3), also known as Machado-Joseph disease (MJD), is an untreatable autosomal dominant neurodegenerative disease, and the most common such inherited ataxia worldwide. The mutation in SCA3 is the expansion of a polymorphic CAG tri-nucleotide repeat sequence in the C-terminal coding region of the ATXN3 gene at chromosomal locus 14q32.1. The mutant ATXN3 protein encoding expanded glutamine (polyQ) sequences interacts with multiple proteins in vivo, and is deposited as aggregates in the SCA3 brain. A large body of literature suggests that the loss of function of the native ATNX3-interacting proteins that are deposited in the polyQ aggregates contributes to cellular toxicity, systemic neurodegeneration and the pathogenic mechanism in SCA3. Nonetheless, a significant understanding of the disease etiology of SCA3, the molecular mechanism by which the polyQ expansions in the mutant ATXN3 induce neurodegeneration in SCA3 has remained elusive. In the present study, we show that the essential DNA strand break repair enzyme PNKP (polynucleotide kinase 3'-phosphatase) interacts with, and is inactivated by, the mutant ATXN3, resulting in inefficient DNA repair, persistent accumulation of DNA damage/strand breaks, and subsequent chronic activation of the DNA damage-response ataxia telangiectasia-mutated (ATM) signaling pathway in SCA3. We report that persistent accumulation of DNA damage/strand breaks and chronic activation of the serine/threonine kinase ATM and the downstream p53 and protein kinase C-δ pro-apoptotic pathways trigger neuronal dysfunction and eventually neuronal death in SCA3. Either PNKP overexpression or pharmacological inhibition of ATM dramatically blocked mutant ATXN3-mediated cell death. Discovery of the mechanism by which mutant ATXN3 induces DNA damage and amplifies the pro-death signaling pathways provides a molecular basis for neurodegeneration due to PNKP inactivation in SCA3, and for the first time offers a possible approach to treatment.
3型脊髓小脑共济失调(SCA3),也称为马查多-约瑟夫病(MJD),是一种无法治愈的常染色体显性神经退行性疾病,也是全球最常见的此类遗传性共济失调。SCA3中的突变是位于染色体位点14q32.1的ATXN3基因C端编码区域中多态性CAG三核苷酸重复序列的扩增。编码扩展谷氨酰胺(polyQ)序列的突变型ATXN3蛋白在体内与多种蛋白质相互作用,并以聚集体形式沉积在SCA3患者的大脑中。大量文献表明,沉积在polyQ聚集体中的天然ATNX3相互作用蛋白功能丧失导致细胞毒性、全身性神经退行性变以及SCA3的致病机制。尽管如此,对于SCA3的疾病病因,即突变型ATXN3中的polyQ扩增诱导SCA3神经退行性变的分子机制,仍不清楚。在本研究中,我们发现必需的DNA链断裂修复酶PNKP(多核苷酸激酶3'-磷酸酶)与突变型ATXN3相互作用并被其灭活,导致DNA修复效率低下、DNA损伤/链断裂持续积累,随后SCA3中DNA损伤反应共济失调毛细血管扩张突变(ATM)信号通路慢性激活。我们报告,DNA损伤/链断裂的持续积累以及丝氨酸/苏氨酸激酶ATM和下游p53及蛋白激酶C-δ促凋亡通路的慢性激活引发SCA3中的神经元功能障碍并最终导致神经元死亡。PNKP过表达或ATM的药理学抑制均可显著阻断突变型ATXN3介导的细胞死亡。突变型ATXN3诱导DNA损伤并放大促死亡信号通路机制的发现为SCA3中PNKP失活导致的神经退行性变提供了分子基础,并首次提供了一种可能的治疗方法。