Ros-Freixedes Roger, Gonen Serap, Gorjanc Gregor, Hickey John M
The Roslin Institute and Royal (Dick) School of Veterinary Studies, The University of Edinburgh, Easter Bush, Midlothian, Scotland, UK.
Genet Sel Evol. 2017 Oct 25;49(1):78. doi: 10.1186/s12711-017-0353-y.
This paper describes a heuristic method for allocating low-coverage sequencing resources by targeting haplotypes rather than individuals. Low-coverage sequencing assembles high-coverage sequence information for every individual by accumulating data from the genome segments that they share with many other individuals into consensus haplotypes. Deriving the consensus haplotypes accurately is critical for achieving a high phasing and imputation accuracy. In order to enable accurate phasing and imputation of sequence information for the whole population, we allocate the available sequencing resources among individuals with existing phased genomic data by targeting the sequencing coverage of their haplotypes.
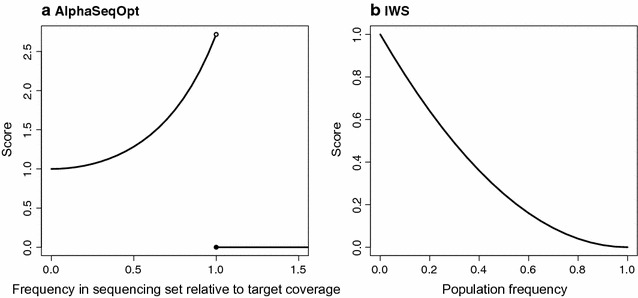
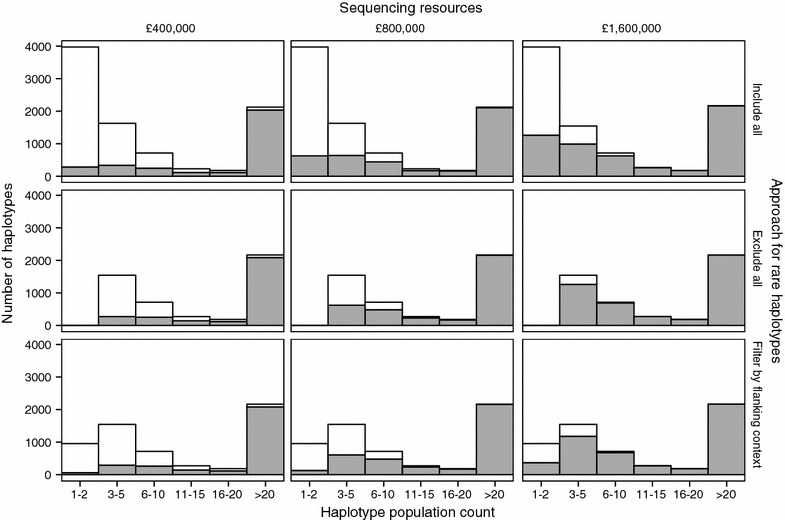
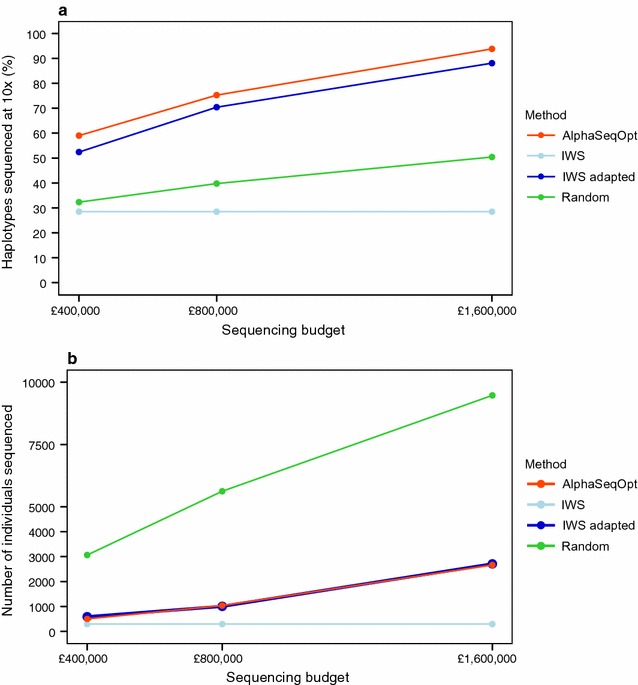
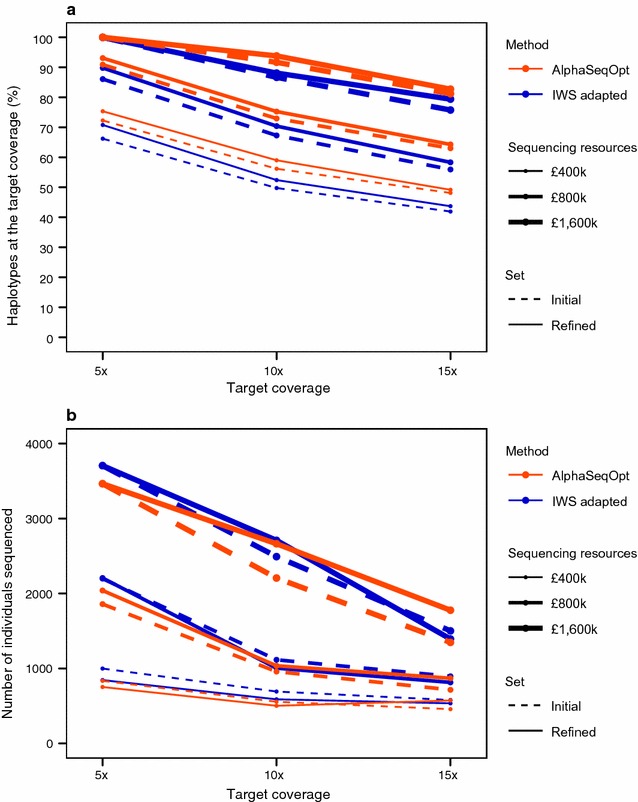
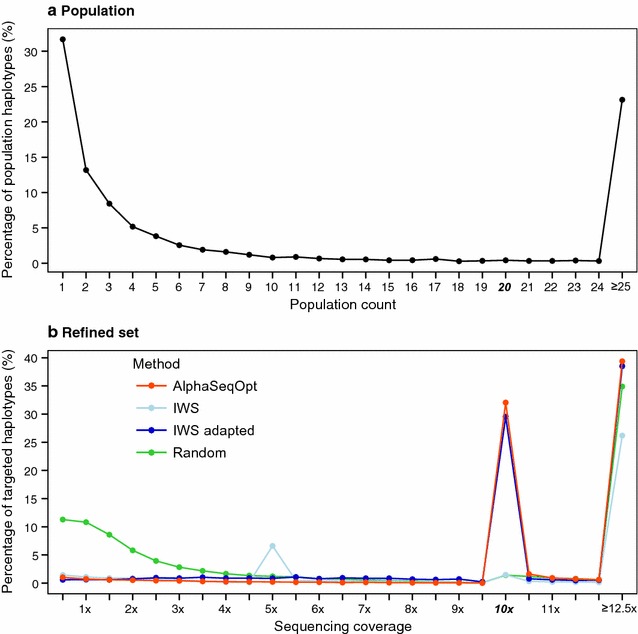
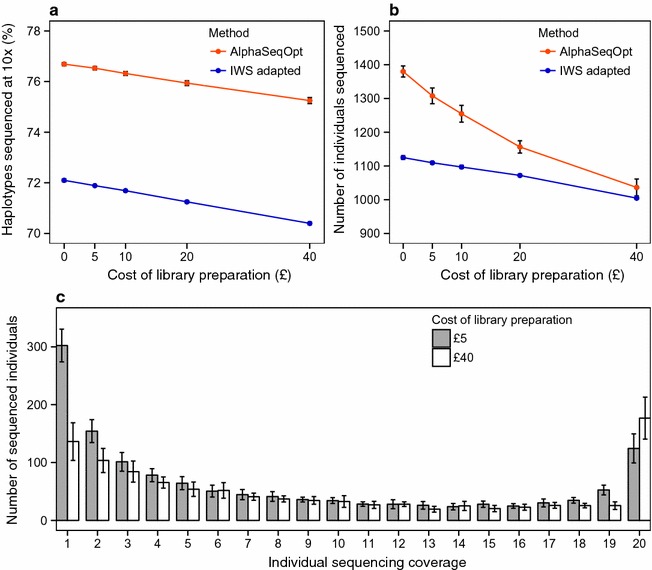
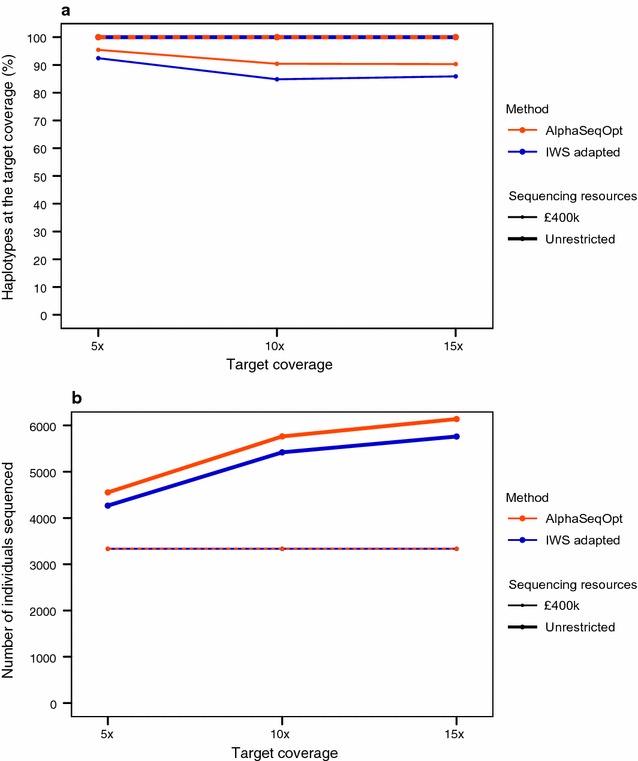
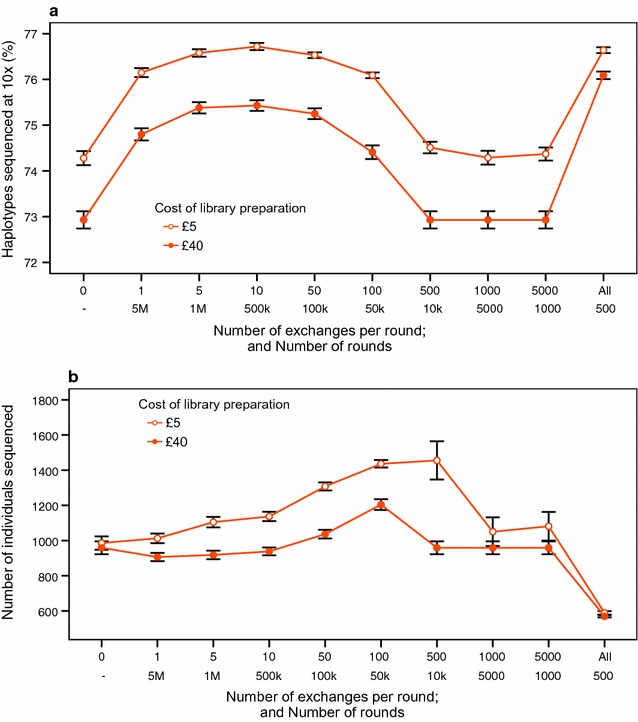
Our method, called AlphaSeqOpt, prioritizes haplotypes using a score function that is based on the frequency of the haplotypes in the sequencing set relative to the target coverage. AlphaSeqOpt has two steps: (1) selection of an initial set of individuals by iteratively choosing the individuals that have the maximum score conditional on the current set, and (2) refinement of the set through several rounds of exchanges of individuals. AlphaSeqOpt is very effective for distributing a fixed amount of sequencing resources evenly across haplotypes, which results in a reduction of the proportion of haplotypes that are sequenced below the target coverage. AlphaSeqOpt can provide a greater proportion of haplotypes sequenced at the target coverage by sequencing less individuals, as compared with other methods that use a score function based on haplotype frequencies in the population. A refinement of the initially selected set can provide a larger more diverse set with more unique individuals, which is beneficial in the context of low-coverage sequencing. We extend the method with an approach for filtering rare haplotypes based on their flanking haplotypes, so that only those that are likely to derive from a recombination event are targeted.
We present a method for allocating sequencing resources so that a greater proportion of haplotypes are sequenced at a coverage that is sufficiently high for population-based imputation with low-coverage sequencing. The haplotype score function, the refinement step, and the new approach for filtering rare haplotypes make AlphaSeqOpt more effective for that purpose than previously reported methods for reducing sequencing redundancy.
本文描述了一种通过针对单倍型而非个体来分配低覆盖度测序资源的启发式方法。低覆盖度测序通过将个体与许多其他个体共享的基因组片段的数据累积为一致单倍型,为每个个体组装高覆盖度序列信息。准确推导一致单倍型对于实现高定相和插补准确性至关重要。为了能够对整个人群的序列信息进行准确的定相和插补,我们通过针对个体单倍型的测序覆盖度,在具有现有定相基因组数据的个体之间分配可用的测序资源。
我们的方法称为AlphaSeqOpt,它使用基于测序集中单倍型频率相对于目标覆盖度的评分函数对单倍型进行优先级排序。AlphaSeqOpt有两个步骤:(1) 通过迭代选择在当前集合条件下得分最高的个体来选择一组初始个体,以及(2) 通过几轮个体交换来优化该集合。AlphaSeqOpt对于将固定量的测序资源均匀分布在单倍型上非常有效,这导致低于目标覆盖度测序的单倍型比例降低。与其他使用基于群体中单倍型频率的评分函数的方法相比,AlphaSeqOpt通过对更少的个体进行测序,可以提供更大比例在目标覆盖度下测序的单倍型。对最初选择的集合进行优化可以提供一个更大、更多样化且包含更多独特个体的集合,这在低覆盖度测序的背景下是有益的。我们通过一种基于侧翼单倍型过滤稀有单倍型的方法扩展了该方法,以便仅针对那些可能源自重组事件的单倍型。
我们提出了一种分配测序资源的方法,以便更大比例的单倍型在足够高的覆盖度下进行测序,从而用于基于群体的低覆盖度测序插补。单倍型评分函数、优化步骤以及过滤稀有单倍型的新方法使AlphaSeqOpt在该目的上比先前报道的减少测序冗余的方法更有效。