The Roslin Institute and Royal (Dick) School of Veterinary Studies, The University of Edinburgh, Midlothian, Scotland, UK.
Genet Sel Evol. 2018 Dec 18;50(1):67. doi: 10.1186/s12711-018-0438-2.
In this paper, we extend multi-locus iterative peeling to provide a computationally efficient method for calling, phasing, and imputing sequence data of any coverage in small or large pedigrees. Our method, called hybrid peeling, uses multi-locus iterative peeling to estimate shared chromosome segments between parents and their offspring at a subset of loci, and then uses single-locus iterative peeling to aggregate genomic information across multiple generations at the remaining loci.
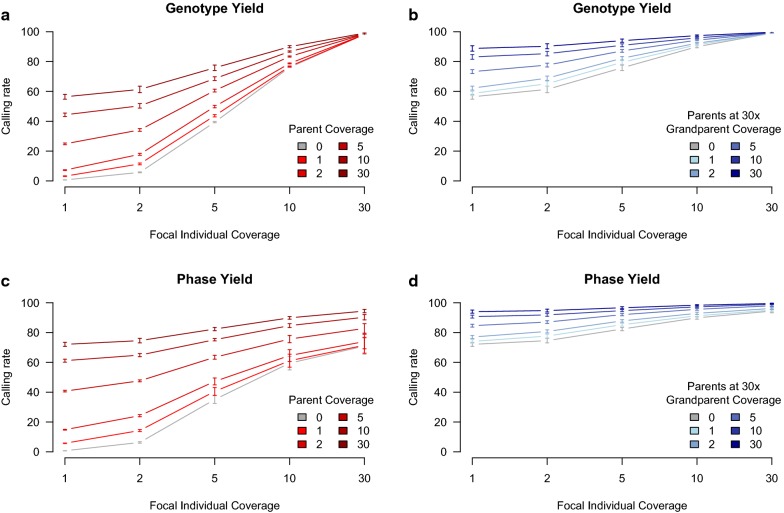
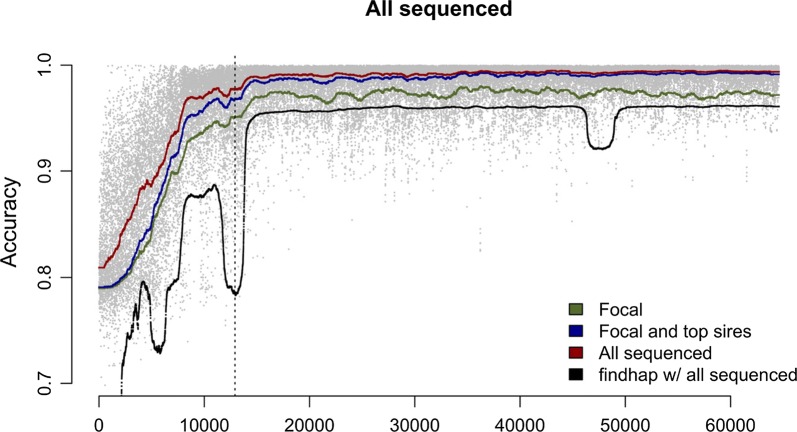
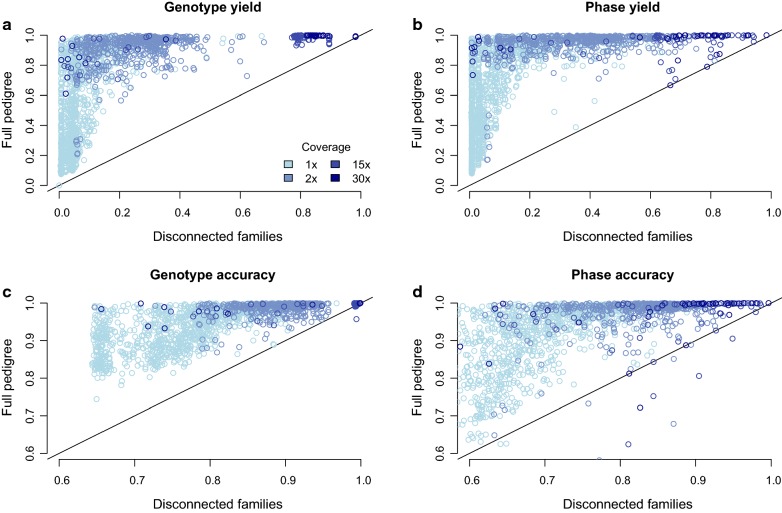
Using a synthetic dataset, we first analysed the performance of hybrid peeling for calling and phasing genotypes in disconnected families, which contained only a focal individual and its parents and grandparents. Second, we analysed the performance of hybrid peeling for calling and phasing genotypes in the context of a full general pedigree. Third, we analysed the performance of hybrid peeling for imputing whole-genome sequence data to non-sequenced individuals in the population. We found that hybrid peeling substantially increased the number of called and phased genotypes by leveraging sequence information on related individuals. The calling rate and accuracy increased when the full pedigree was used compared to a reduced pedigree of just parents and grandparents. Finally, hybrid peeling imputed accurately whole-genome sequence to non-sequenced individuals.
We believe that this algorithm will enable the generation of low cost and high accuracy whole-genome sequence data in many pedigreed populations. We make this algorithm available as a standalone program called AlphaPeel.
在本文中,我们扩展了多基因座迭代剥除法,提供了一种计算效率高的方法,用于在小或大家系中调用、定相和插入任何覆盖度的序列数据。我们的方法称为混合剥除法,它使用多基因座迭代剥除法在一组基因座上估计父母与其后代之间的共享染色体片段,然后使用单基因座迭代剥除法在其余基因座上聚合多个世代的基因组信息。
使用合成数据集,我们首先分析了混合剥除法在分离家族(仅包含一个焦点个体及其父母和祖父母)中进行基因型调用和定相的性能。其次,我们分析了混合剥除法在全一般家系中进行基因型调用和定相的性能。第三,我们分析了混合剥除法在向人群中未测序个体中插入全基因组序列数据的性能。我们发现,混合剥除法通过利用相关个体的序列信息,大大增加了调用和定相基因型的数量。与仅父母和祖父母的简化家谱相比,使用完整家谱时,调用率和准确性提高了。最后,混合剥除法准确地向未测序个体插入了全基因组序列。
我们相信,这种算法将能够在许多有谱系的人群中生成低成本、高精度的全基因组序列数据。我们将这个算法作为一个独立的程序称为 AlphaPeel 提供。