Department of Genetics, University of São Paulo, São Paulo 13418-900, Brazil.
Department of Horticultural Sciences, Texas A&M University, College Station, TX 77843-0001, USA.
Gigascience. 2022 Dec 28;12. doi: 10.1093/gigascience/giad092. Epub 2023 Oct 27.
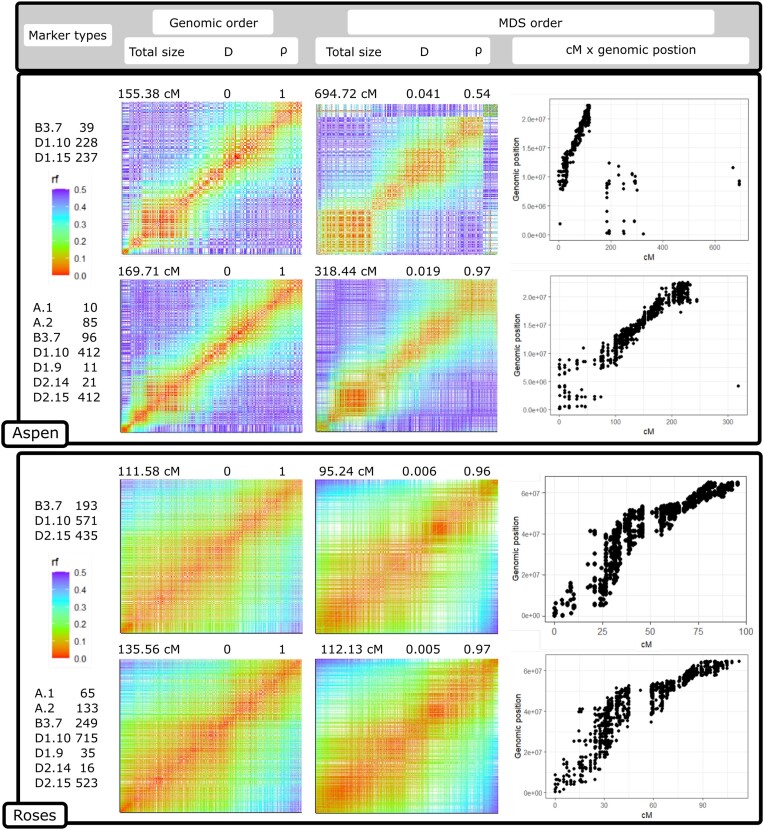
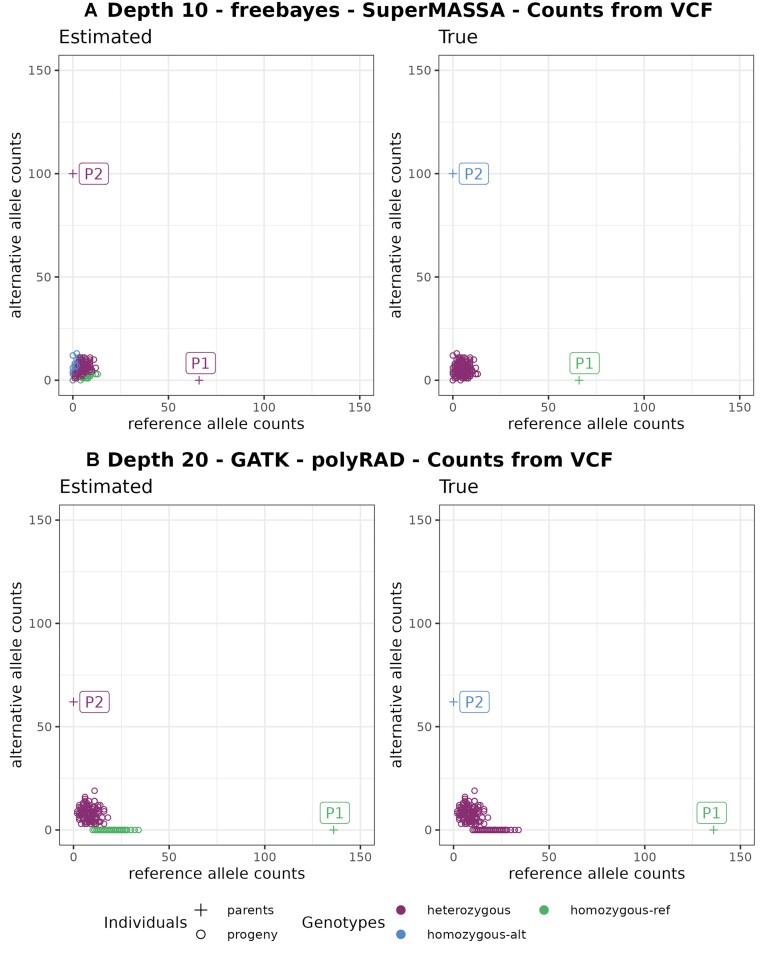
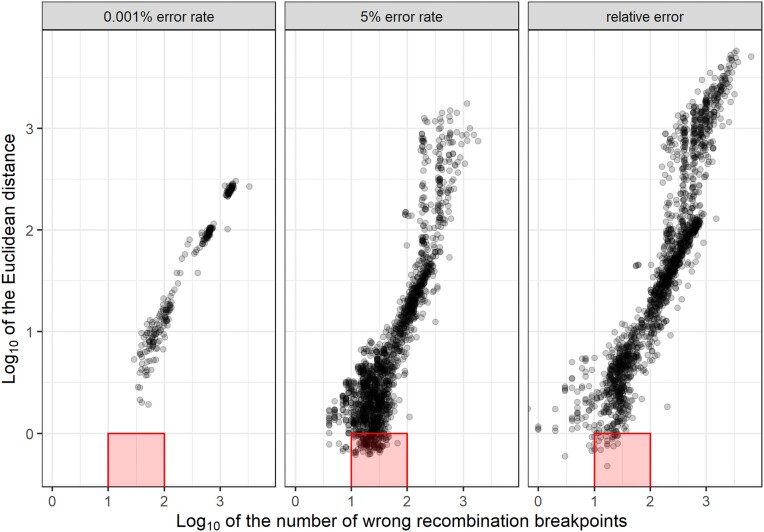
Genotyping-by-sequencing (GBS) provides affordable methods for genotyping hundreds of individuals using millions of markers. However, this challenges bioinformatic procedures that must overcome possible artifacts such as the bias generated by polymerase chain reaction duplicates and sequencing errors. Genotyping errors lead to data that deviate from what is expected from regular meiosis. This, in turn, leads to difficulties in grouping and ordering markers, resulting in inflated and incorrect linkage maps. Therefore, genotyping errors can be easily detected by linkage map quality evaluations.
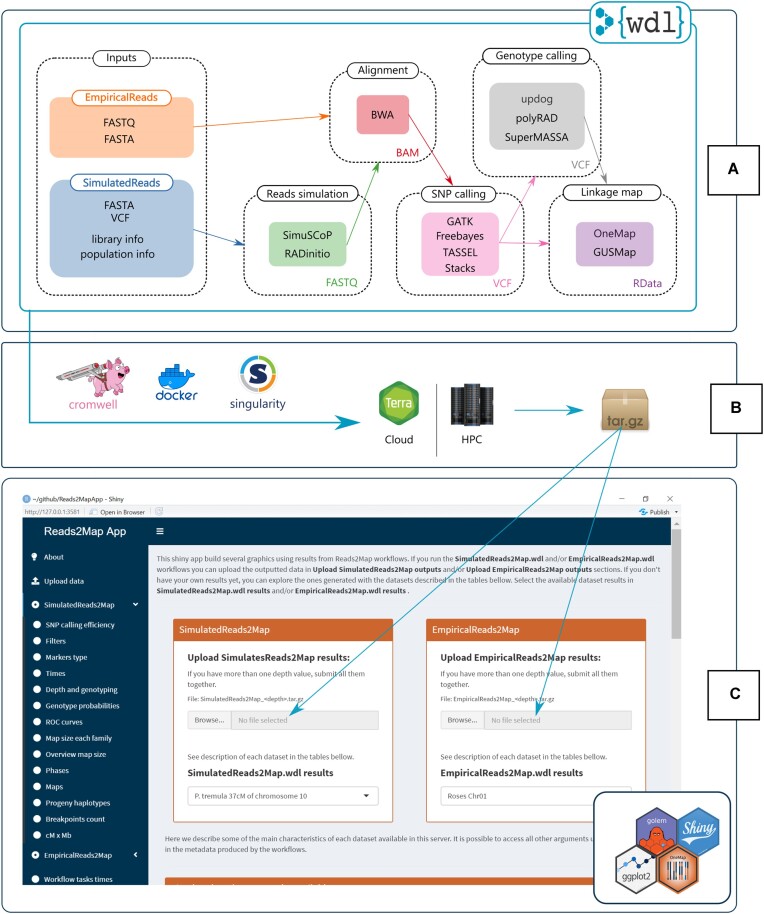
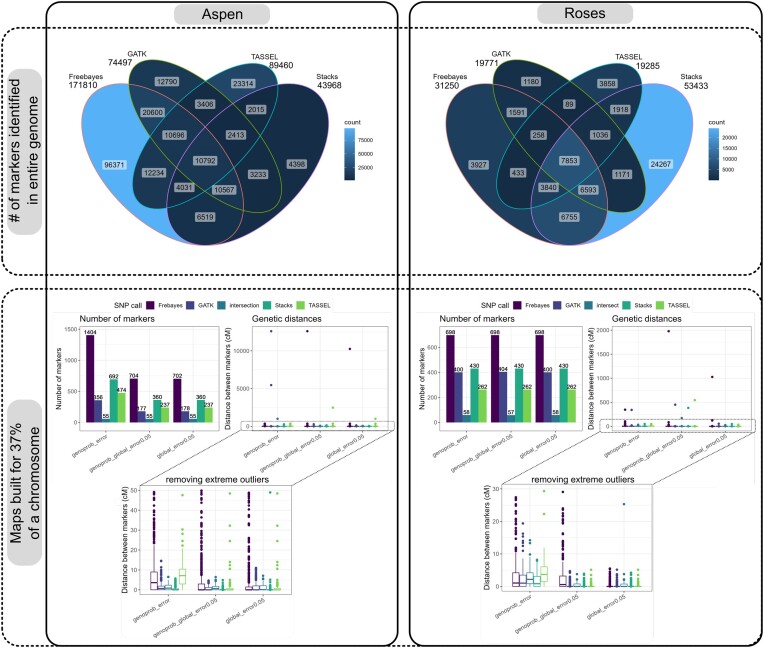
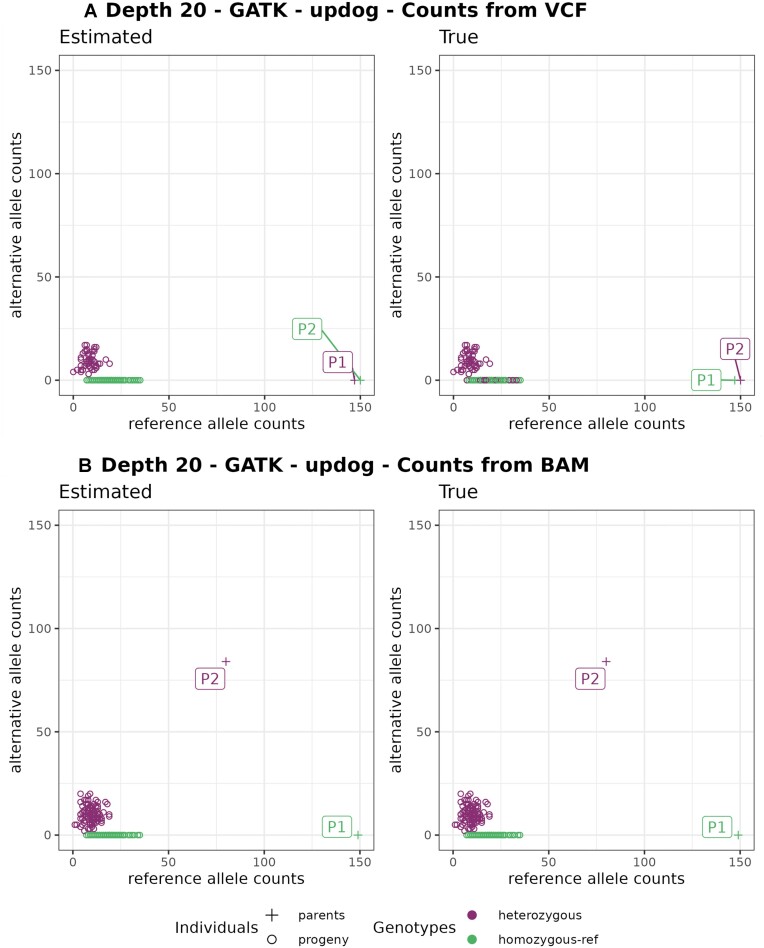
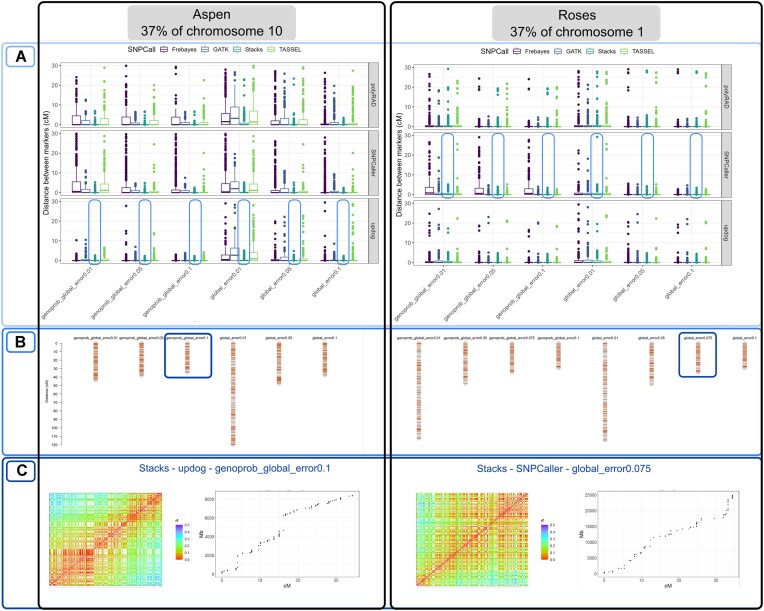
We developed and used the Reads2Map workflow to build linkage maps with simulated and empirical GBS data of diploid outcrossing populations. The workflows run GATK, Stacks, TASSEL, and Freebayes for single-nucleotide polymorphism calling and updog, polyRAD, and SuperMASSA for genotype calling, as well as OneMap and GUSMap to build linkage maps. Using simulated data, we observed which genotype call software fails in identifying common errors in GBS sequencing data and proposed specific filters to better handle them. We tested whether it is possible to overcome errors in a linkage map using genotype probabilities from each software or global error rates to estimate genetic distances with an updated version of OneMap. We also evaluated the impact of segregation distortion, contaminant samples, and haplotype-based multiallelic markers in the final linkage maps. Through our evaluations, we observed that some of the approaches produce different results depending on the dataset (dataset dependent) and others produce consistent advantageous results among them (dataset independent).
We set as default in the Reads2Map workflows the approaches that showed to be dataset independent for GBS datasets according to our results. This reduces the number of required tests to identify optimal pipelines and parameters for other empirical datasets. Using Reads2Map, users can select the pipeline and parameters that best fit their data context. The Reads2MapApp shiny app provides a graphical representation of the results to facilitate their interpretation.
测序基因分型(GBS)提供了一种经济实惠的方法,可使用数百万个标记对数百个人进行基因分型。然而,这给生物信息学程序带来了挑战,这些程序必须克服聚合酶链反应重复和测序错误等可能产生的伪影。基因分型错误导致的数据偏离了常规减数分裂所预期的数据。这反过来又导致标记的分组和排序困难,从而导致连锁图谱膨胀和错误。因此,基因分型错误可以通过连锁图谱质量评估轻松检测。
我们开发并使用了 Reads2Map 工作流程,使用二倍体杂交群体的模拟和经验 GBS 数据构建连锁图谱。工作流程运行 GATK、Stacks、TASSEL 和 Freebayes 进行单核苷酸多态性调用,updog、polyRAD 和 SuperMASSA 进行基因型调用,以及 OneMap 和 GUSMap 进行连锁图谱构建。使用模拟数据,我们观察到哪种基因型调用软件无法识别 GBS 测序数据中的常见错误,并提出了特定的筛选器来更好地处理这些错误。我们测试了是否可以使用每个软件的基因型概率或全局错误率来克服连锁图谱中的错误,以使用 OneMap 的更新版本估计遗传距离。我们还评估了分离失真、污染样本和基于单倍型的多等位基因标记对最终连锁图谱的影响。通过我们的评估,我们观察到一些方法根据数据集产生不同的结果(数据集依赖),而另一些方法在它们之间产生一致的有利结果(数据集独立)。
根据我们的结果,我们在 Reads2Map 工作流程中将默认设置为那些针对 GBS 数据集表现为数据集独立的方法。这减少了识别其他经验数据集最佳管道和参数所需的测试数量。使用 Reads2Map,用户可以选择最适合其数据上下文的管道和参数。Reads2MapApp shiny 应用程序提供了结果的图形表示,以方便解释。