Brain Tumor Center, Cincinnati Children's Hospital Medical Center, 3333 Burnet Avenue, Cincinnati, OH, 45229, USA.
Department of Human Genetics, McGill University, Montreal, QC, H3A 1B1, Canada.
Acta Neuropathol Commun. 2017 Oct 30;5(1):78. doi: 10.1186/s40478-017-0479-8.
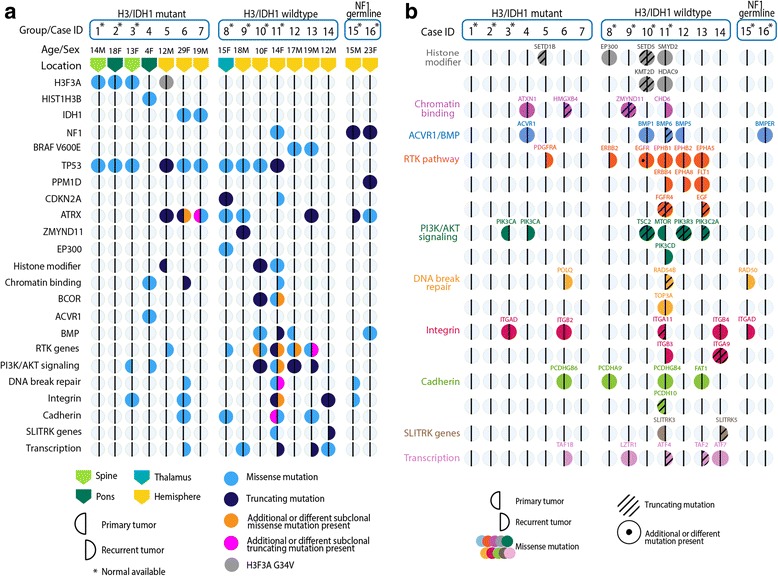
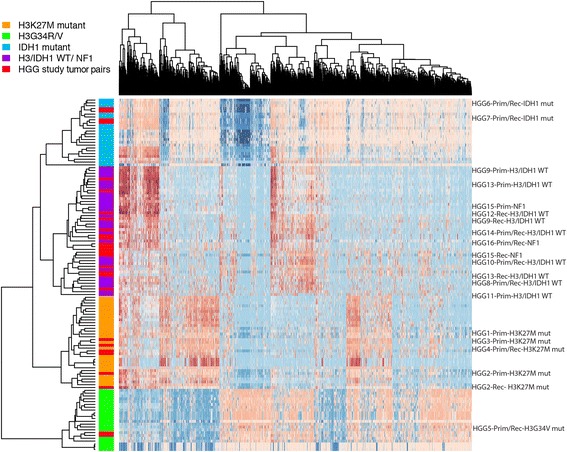
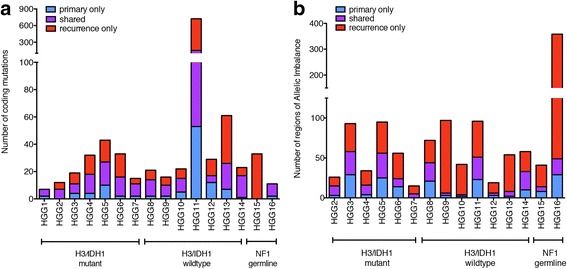
Pediatric high-grade gliomas (pHGGs) are aggressive neoplasms representing approximately 20% of brain tumors in children. Current therapies offer limited disease control, and patients have a poor prognosis. Empiric use of targeted therapy, especially at progression, is increasingly practiced despite a paucity of data regarding temporal and therapy-driven genomic evolution in pHGGs. To study the genetic landscape of pHGGs at recurrence, we performed whole exome and methylation analyses on matched primary and recurrent pHGGs from 16 patients. Tumor mutational profiles identified three distinct subgroups. Group 1 (n = 7) harbored known hotspot mutations in Histone 3 (H3) (K27M or G34V) or IDH1 (H3/IDH1 mutants) and co-occurring TP53 or ACVR1 mutations in tumor pairs across the disease course. Group 2 (n = 7), H3/IDH1 wildtype tumor pairs, harbored novel mutations in chromatin modifiers (ZMYND11, EP300 n = 2), all associated with TP53 alterations, or had BRAF V600E mutations (n = 2) conserved across tumor pairs. Group 3 included 2 tumors with NF1 germline mutations. Pairs from primary and relapsed pHGG samples clustered within the same DNA methylation subgroup. ATRX mutations were clonal and retained in H3G34V and H3/IDH1 wildtype tumors, while different genetic alterations in this gene were observed at diagnosis and recurrence in IDH1 mutant tumors. Mutations in putative drug targets (EGFR, ERBB2, PDGFRA, PI3K) were not always shared between primary and recurrence samples, indicating evolution during progression. Our findings indicate that specific key driver mutations in pHGGs are conserved at recurrence and are prime targets for therapeutic development and clinical trials (e.g. H3 post-translational modifications, IDH1, BRAF V600E). Other actionable mutations are acquired or lost, indicating that re-biopsy at recurrence will provide better guidance for effective targeted therapy of pHGGs.
儿科高级别神经胶质瘤(pHGG)是侵袭性肿瘤,约占儿童脑肿瘤的 20%。目前的治疗方法对疾病的控制效果有限,患儿预后较差。尽管在 pHGG 中,关于时间和治疗驱动的基因组进化的数据很少,但靶向治疗的经验性应用,尤其是在进展时,越来越普遍。为了研究 pHGG 复发时的遗传特征,我们对 16 名患者的配对原发和复发 pHGG 进行了全外显子组和甲基化分析。肿瘤突变图谱确定了三个不同的亚组。第 1 组(n=7)在疾病过程中,肿瘤对中存在已知的组蛋白 3(H3)(K27M 或 G34V)或 IDH1(H3/IDH1 突变体)热点突变,以及 TP53 或 ACVR1 突变。第 2 组(n=7),H3/IDH1 野生型肿瘤对,存在染色质修饰因子(ZMYND11、EP300,各 2 例)的新突变,均与 TP53 改变相关,或存在 BRAF V600E 突变(2 例),在肿瘤对中均保持一致。第 3 组包括 2 例 NF1 种系突变肿瘤。原发性和复发性 pHGG 样本的配对聚类在同一 DNA 甲基化亚组中。ATRX 突变是克隆的,在 H3G34V 和 H3/IDH1 野生型肿瘤中保留,而在 IDH1 突变型肿瘤中,该基因的不同遗传改变在诊断和复发时都存在。潜在药物靶点(EGFR、ERBB2、PDGFRA、PI3K)的突变并非总是在原发性和复发性样本中共享,表明在进展过程中发生了进化。我们的研究结果表明,pHGG 中的特定关键驱动突变在复发时是保守的,是开发治疗方法和临床试验的主要目标(例如 H3 翻译后修饰、IDH1、BRAF V600E)。其他可操作的突变是获得或丢失的,这表明在复发时进行再次活检将为 pHGG 的有效靶向治疗提供更好的指导。