Carcelén María, Abascal Estefanía, Herranz Marta, Santantón Sheila, Zenteno Roberto, Ruiz Serrano María Jesús, Bouza Emilio, Pérez-Lago Laura, García-de-Viedma Darío
Servicio de Microbiología Clínica y Enfermedades Infecciosas, Hospital General Universitario Gregorio Marañón, Madrid, Spain.
Instituto de Investigación Sanitaria Gregorio Marañón, Madrid, Spain.
PLoS One. 2017 Nov 1;12(11):e0186956. doi: 10.1371/journal.pone.0186956. eCollection 2017.
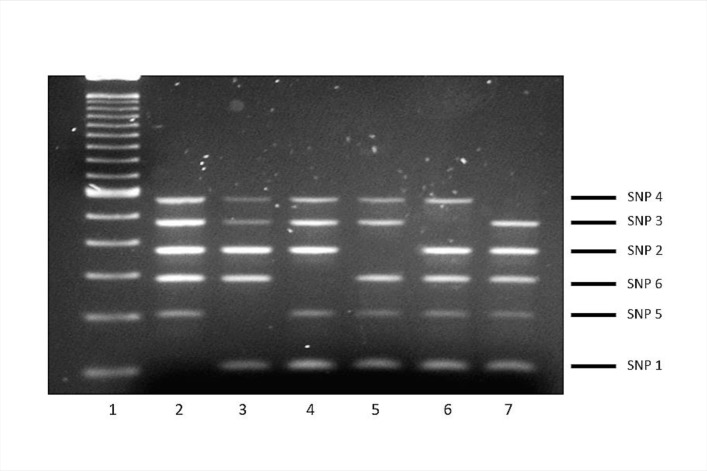
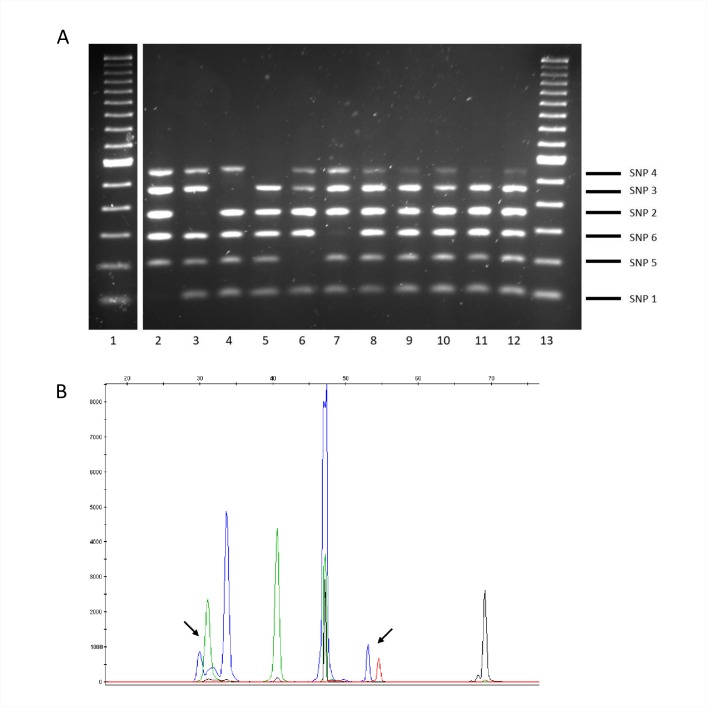
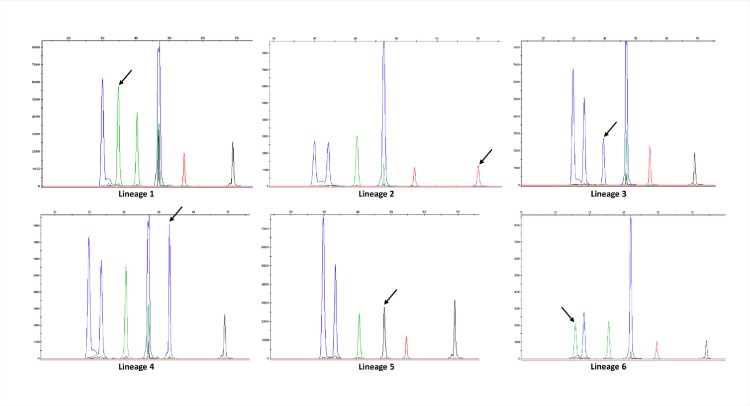
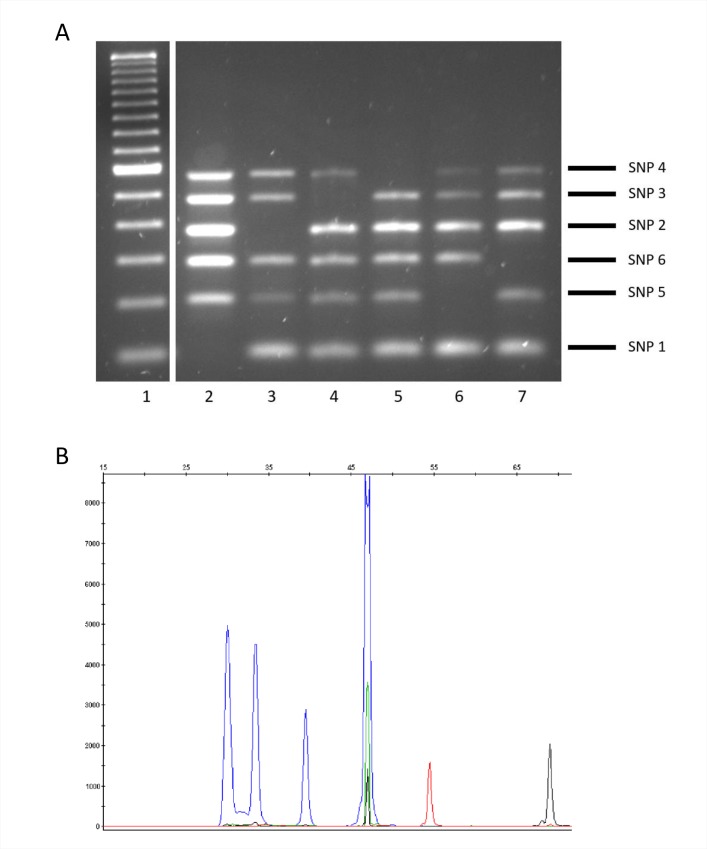
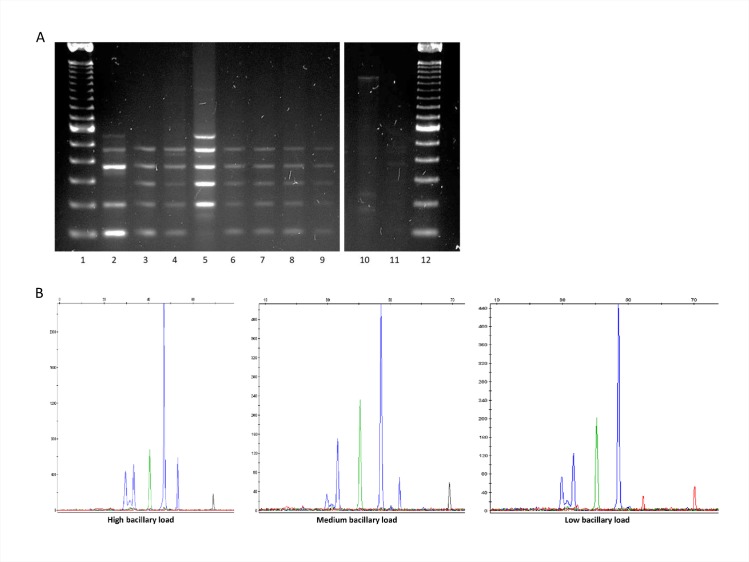
The assignation of lineages in Mycobacterium tuberculosis (MTB) provides valuable information for evolutionary and phylogeographic studies and makes for more accurate knowledge of the distribution of this pathogen worldwide. Differences in virulence have also been found for certain lineages. MTB isolates were initially assigned to lineages based on data obtained from genotyping techniques, such as spoligotyping or MIRU-VNTR analysis, some of which are more suitable for molecular epidemiology studies. However, since these methods are subject to a certain degree of homoplasy, other criteria have been chosen to assign lineages. These are based on targeting robust and specific SNPs for each lineage. Here, we propose two newly designed multiplex targeting methods-both of which are single-tube tests-to optimize the assignation of the six main lineages in MTB. The first method is based on ASO-PCR and offers an inexpensive and easy-to-implement assay for laboratories with limited resources. The other, which is based on SNaPshot, enables more refined standardized assignation of lineages for laboratories with better resources. Both methods performed well when assigning lineages from cultured isolates from a control panel, a test panel, and a problem panel from an unrelated population, Mexico, which included isolates in which standard genotyping was not able to classify lineages. Both tests were also able to assign lineages from stored isolates, without the need for subculture or purification of DNA, and even directly from clinical specimens with a medium-high bacilli burden. Our assays could broaden the contexts where information on lineages can be acquired, thus enabling us to quickly update data from retrospective collections and to merge data with those obtained at the time of diagnosis of a new TB case.
结核分枝杆菌(MTB)谱系的划分可为进化和系统地理学研究提供有价值的信息,有助于更准确地了解这种病原体在全球的分布情况。某些谱系的毒力也存在差异。MTB分离株最初是根据基因分型技术(如间隔寡核苷酸分型或MIRU-VNTR分析)获得的数据来划分谱系的,其中一些技术更适合分子流行病学研究。然而,由于这些方法存在一定程度的同塑性,因此选择了其他标准来划分谱系。这些标准基于针对每个谱系的稳健且特异的单核苷酸多态性(SNP)。在此,我们提出了两种新设计的多重靶向方法——均为单管检测——以优化MTB六个主要谱系的划分。第一种方法基于等位基因特异性寡核苷酸PCR(ASO-PCR),为资源有限的实验室提供了一种廉价且易于实施的检测方法。另一种基于SNaPshot,可为资源更丰富的实验室实现更精细的谱系标准化划分。当对来自对照组、测试组和来自无关人群(墨西哥)的问题组的培养分离株进行谱系划分时,这两种方法都表现良好,其中包括标准基因分型无法分类谱系的分离株。这两种检测方法还能够对储存的分离株进行谱系划分,无需进行传代培养或DNA纯化,甚至可以直接从具有中高菌量的临床标本中进行划分。我们的检测方法可以拓宽获取谱系信息的背景,从而使我们能够快速更新回顾性收集的数据,并将这些数据与新结核病病例诊断时获得的数据合并。