Department of Computer Science and Engineering, United International University, House 80, Road 8A, Dhanmondi, Dhaka, 1209, Bangladesh.
Department of Computer Science, Morgan State University, Baltimore, Maryland, United States.
Sci Rep. 2017 Nov 2;7(1):14938. doi: 10.1038/s41598-017-14945-1.
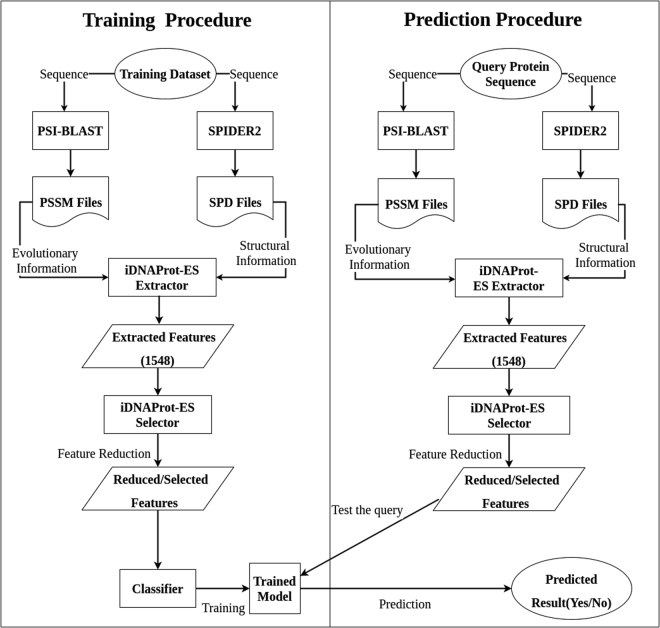
DNA-binding proteins play a very important role in the structural composition of the DNA. In addition, they regulate and effect various cellular processes like transcription, DNA replication, DNA recombination, repair and modification. The experimental methods used to identify DNA-binding proteins are expensive and time consuming and thus attracted researchers from computational field to address the problem. In this paper, we present iDNAProt-ES, a DNA-binding protein prediction method that utilizes both sequence based evolutionary and structure based features of proteins to identify their DNA-binding functionality. We used recursive feature elimination to extract an optimal set of features and train them using Support Vector Machine (SVM) with linear kernel to select the final model. Our proposed method significantly outperforms the existing state-of-the-art predictors on standard benchmark dataset. The accuracy of the predictor is 90.18% using jack knife test and 88.87% using 10-fold cross validation on the benchmark dataset. The accuracy of the predictor on the independent dataset is 80.64% which is also significantly better than the state-of-the-art methods. iDNAProt-ES is a novel prediction method that uses evolutionary and structural based features. We believe the superior performance of iDNAProt-ES will motivate the researchers to use this method to identify DNA-binding proteins. iDNAProt-ES is publicly available as a web server at: http://brl.uiu.ac.bd/iDNAProt-ES/ .
DNA 结合蛋白在 DNA 的结构组成中起着非常重要的作用。此外,它们还调节和影响转录、DNA 复制、DNA 重组、修复和修饰等各种细胞过程。用于鉴定 DNA 结合蛋白的实验方法既昂贵又耗时,因此吸引了计算领域的研究人员来解决这个问题。在本文中,我们提出了 iDNAProt-ES,这是一种 DNA 结合蛋白预测方法,它利用蛋白质的序列进化和结构特征来识别其 DNA 结合功能。我们使用递归特征消除来提取最佳特征集,并使用带有线性核的支持向量机 (SVM) 对其进行训练,以选择最终模型。我们提出的方法在标准基准数据集上显著优于现有的最先进的预测器。在基准数据集上,使用 jack knife 测试和 10 倍交叉验证的预测器的准确率分别为 90.18%和 88.87%。在独立数据集上的预测器的准确率为 80.64%,也明显优于最先进的方法。iDNAProt-ES 是一种使用进化和基于结构的特征的新型预测方法。我们相信 iDNAProt-ES 的优越性能将激励研究人员使用这种方法来鉴定 DNA 结合蛋白。iDNAProt-ES 作为一个网络服务器,可在以下网址获得:http://brl.uiu.ac.bd/iDNAProt-ES/ 。