European Research Institute for the Biology of Ageing, University Medical Center Groningen, University of Groningen, Building 3226, 9713 AV, Groningen, The Netherlands.
Max Planck Institute for Informatics, Saarbrücken, Germany.
Nat Commun. 2017 Nov 3;8(1):1293. doi: 10.1038/s41467-017-01389-4.
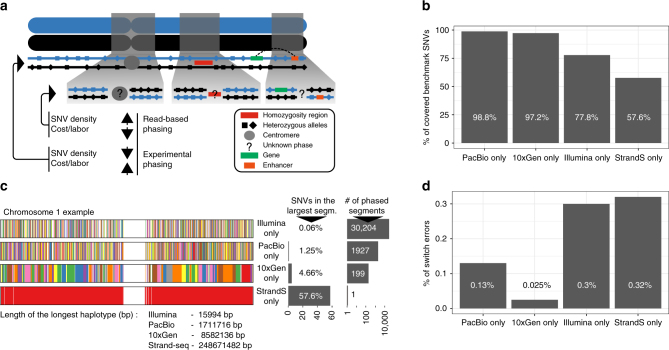
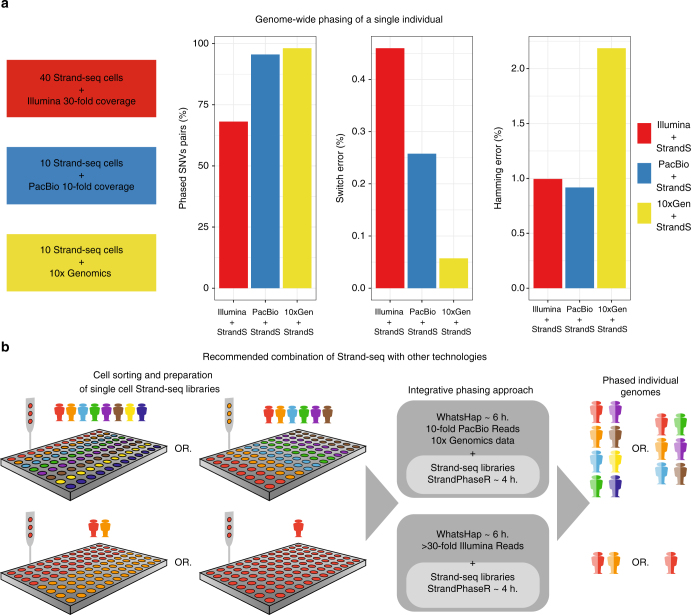
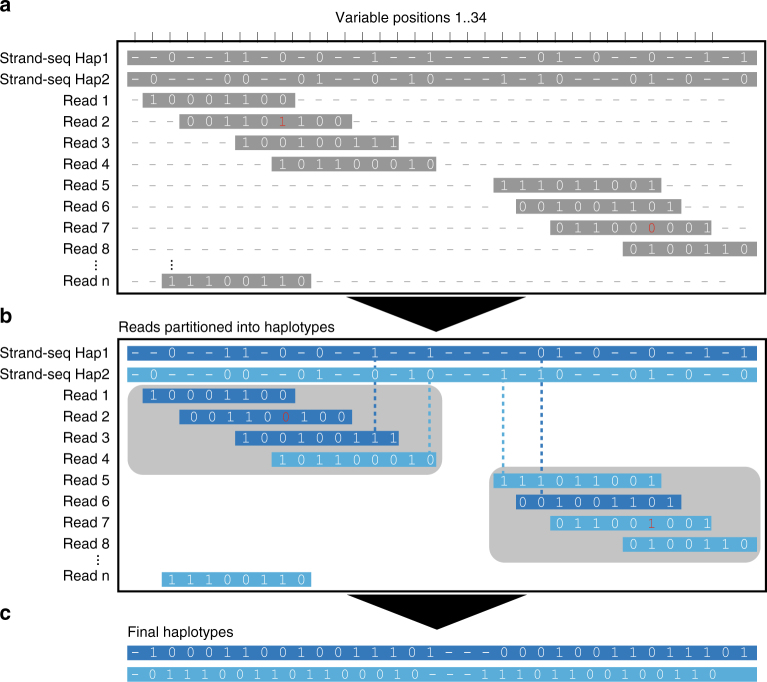
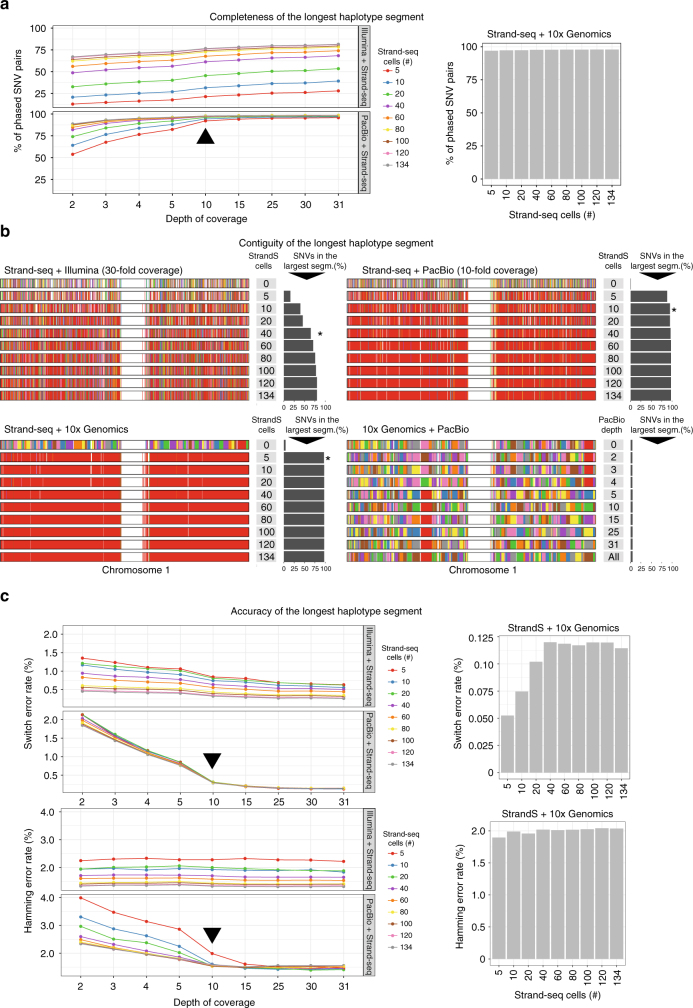
The diploid nature of the human genome is neglected in many analyses done today, where a genome is perceived as a set of unphased variants with respect to a reference genome. This lack of haplotype-level analyses can be explained by a lack of methods that can produce dense and accurate chromosome-length haplotypes at reasonable costs. Here we introduce an integrative phasing strategy that combines global, but sparse haplotypes obtained from strand-specific single-cell sequencing (Strand-seq) with dense, yet local, haplotype information available through long-read or linked-read sequencing. We provide comprehensive guidance on the required sequencing depths and reliably assign more than 95% of alleles (NA12878) to their parental haplotypes using as few as 10 Strand-seq libraries in combination with 10-fold coverage PacBio data or, alternatively, 10X Genomics linked-read sequencing data. We conclude that the combination of Strand-seq with different technologies represents an attractive solution to chart the genetic variation of diploid genomes.
在今天进行的许多分析中,人类基因组的二倍体性质被忽视了,在这些分析中,基因组被视为相对于参考基因组的一组未分相变体。这种缺乏单倍型水平分析的情况可以解释为缺乏能够以合理的成本生成密集且准确的染色体长度单倍型的方法。在这里,我们介绍了一种综合的相位策略,该策略将从链特异性单细胞测序(Strand-seq)获得的全局但稀疏的单倍型与通过长读长或连接读测序获得的密集但局部的单倍型信息相结合。我们提供了全面的测序深度指导,使用仅 10 个 Strand-seq 文库与 10 倍覆盖度的 PacBio 数据相结合,或者使用 10X Genomics 连接读测序数据,可靠地将超过 95%的等位基因(NA12878)分配给它们的亲本单倍型。我们得出结论,Strand-seq 与不同技术的结合代表了绘制二倍体基因组遗传变异的一种有吸引力的解决方案。