Nigatu Dawit, Sobetzko Patrick, Yousef Malik, Henkel Werner
Transmission Systems Group, Jacobs University Bremen, Campus Ring 1, Bremen, D-28759, Germany.
Philipps-Universität Marburg, LOEWE-Zentrum für Synthetische Mikrobiologie, Hans-Meerwein-Straße, Mehrzweckgebäude, Marburg, 35043, Germany.
BMC Bioinformatics. 2017 Nov 9;18(1):473. doi: 10.1186/s12859-017-1884-5.
Identification of essential genes is not only useful for our understanding of the minimal gene set required for cellular life but also aids the identification of novel drug targets in pathogens. In this work, we present a simple and effective gene essentiality prediction method using information-theoretic features that are derived exclusively from the gene sequences.
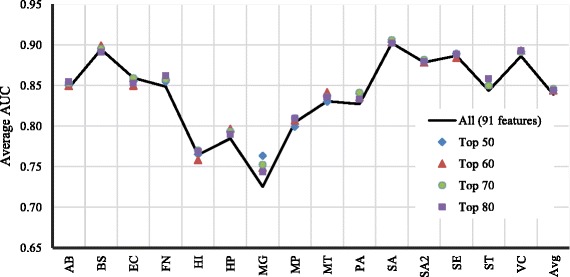
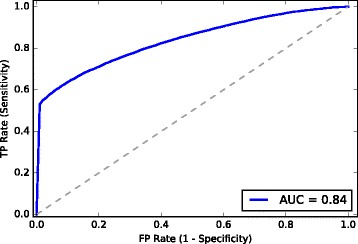
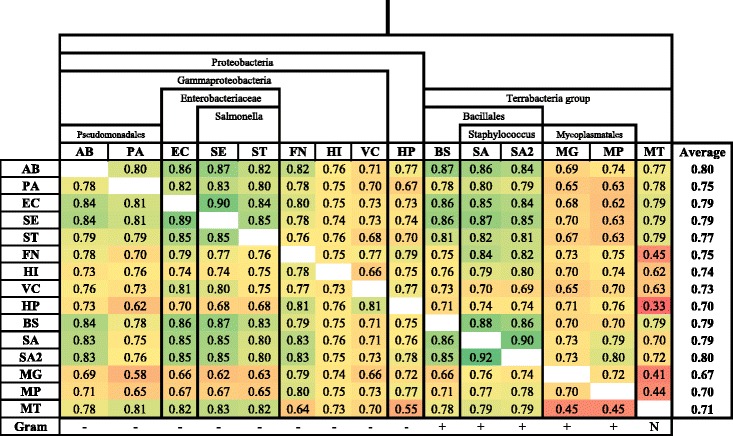
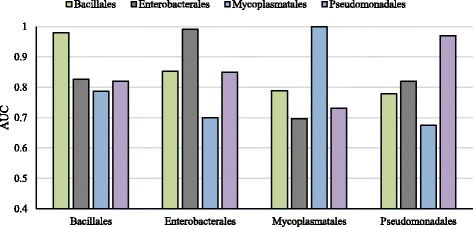
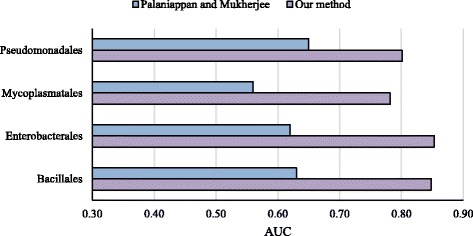
We developed a Random Forest classifier and performed an extensive model performance evaluation among and within 15 selected bacteria. In intra-organism predictions, where training and testing sets are taken from the same organism, AUC (Area Under the Curve) scores ranging from 0.73 to 0.90, 0.84 on average, were obtained. Cross-organism predictions using 5-fold cross-validation, pairwise, leave-one-species-out, leave-one-taxon-out, and cross-taxon yielded average AUC scores of 0.88, 0.75, 0.80, 0.82, and 0.78, respectively. To further show the applicability of our method in other domains of life, we predicted the essential genes of the yeast Schizosaccharomyces pombe and obtained a similar accuracy (AUC 0.84).
The proposed method enables a simple and reliable identification of essential genes without searching in databases for orthologs and demanding further experimental data such as network topology and gene-expression.
鉴定必需基因不仅有助于我们理解细胞生命所需的最小基因集,还有助于识别病原体中的新型药物靶点。在这项工作中,我们提出了一种简单有效的基因必需性预测方法,该方法使用仅从基因序列中导出的信息论特征。
我们开发了一种随机森林分类器,并在15种选定的细菌内部和之间进行了广泛的模型性能评估。在生物体内部预测中,训练集和测试集取自同一生物体,获得的曲线下面积(AUC)分数范围为0.73至0.90,平均为0.84。使用5折交叉验证、成对、留一物种法、留一分类单元法和交叉分类单元法进行的跨生物体预测,平均AUC分数分别为0.88、0.75、0.80、0.82和0.78。为了进一步展示我们的方法在生命其他领域的适用性,我们预测了粟酒裂殖酵母的必需基因,并获得了相似的准确率(AUC 0.84)。
所提出的方法能够简单可靠地鉴定必需基因,而无需在数据库中搜索直系同源物,也不需要诸如网络拓扑和基因表达等进一步的实验数据。