Campos Tulio L, Korhonen Pasi K, Gasser Robin B, Young Neil D
Department of Veterinary Biosciences, Melbourne Veterinary School, The University of Melbourne, Parkville, Victoria 3010, Australia.
Bioinformatics Core Facility, Instituto Aggeu Magalhães, Fundação Oswaldo Cruz (IAM-Fiocruz), Recife, Pernambuco, Brazil.
Comput Struct Biotechnol J. 2019 Jun 8;17:785-796. doi: 10.1016/j.csbj.2019.05.008. eCollection 2019.
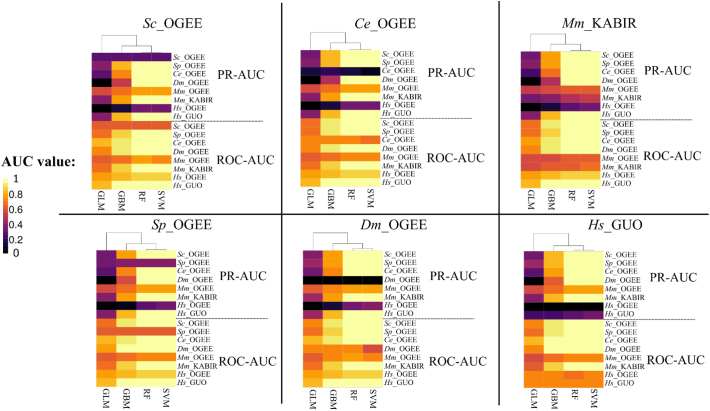
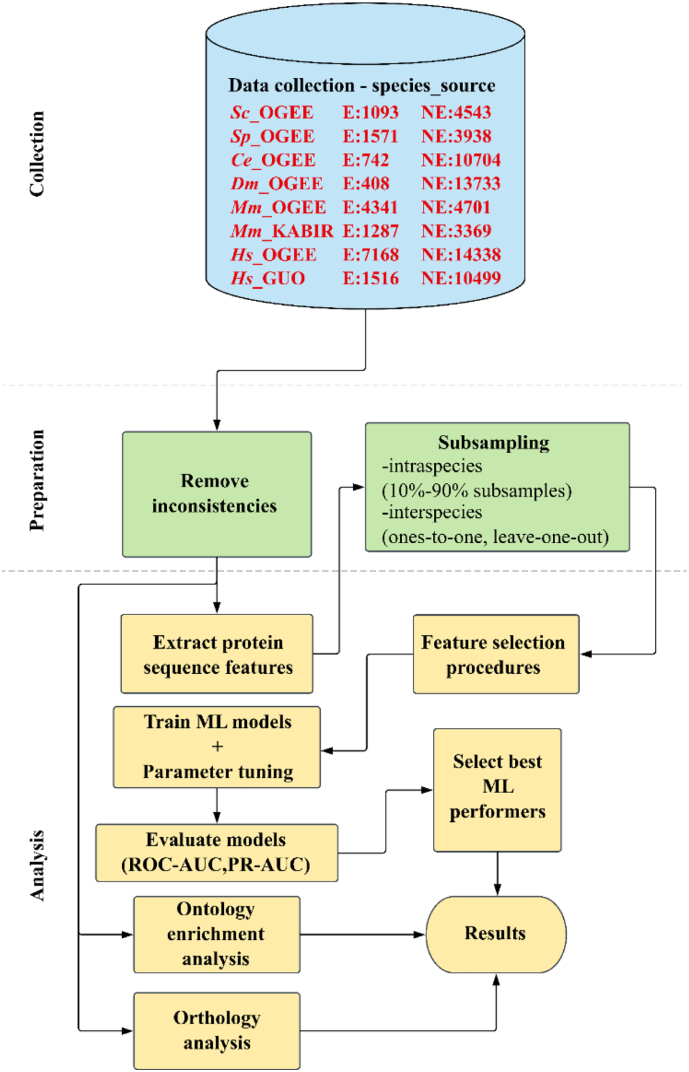
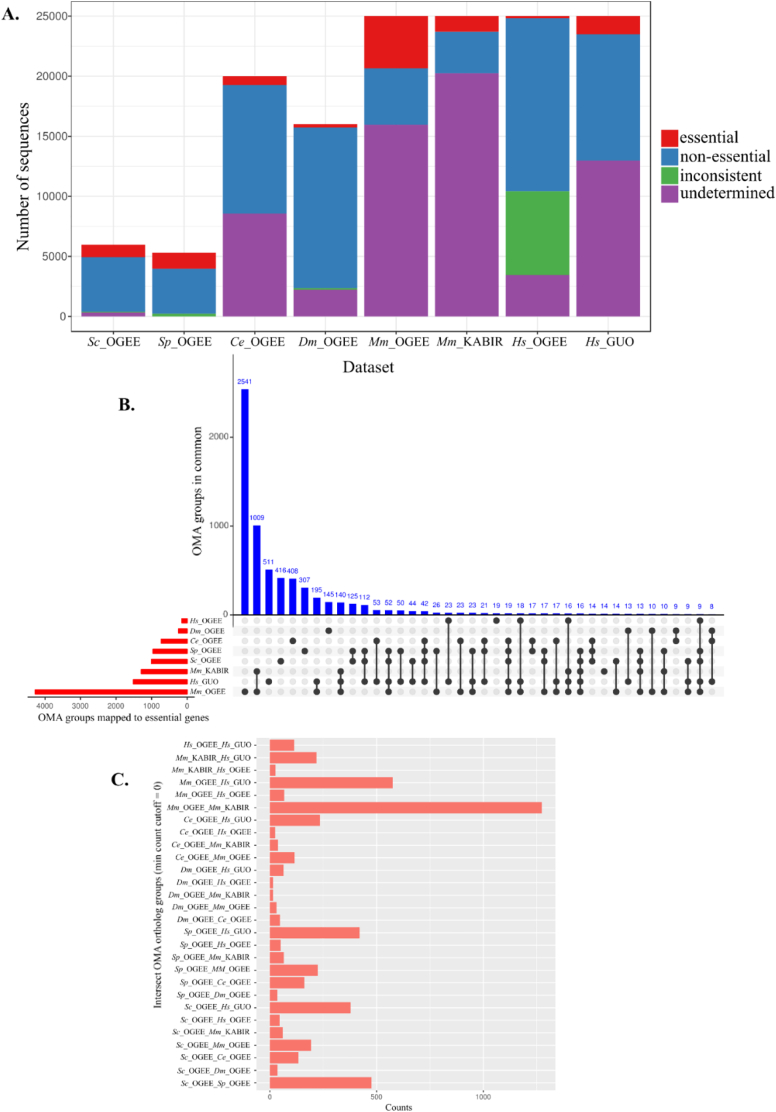
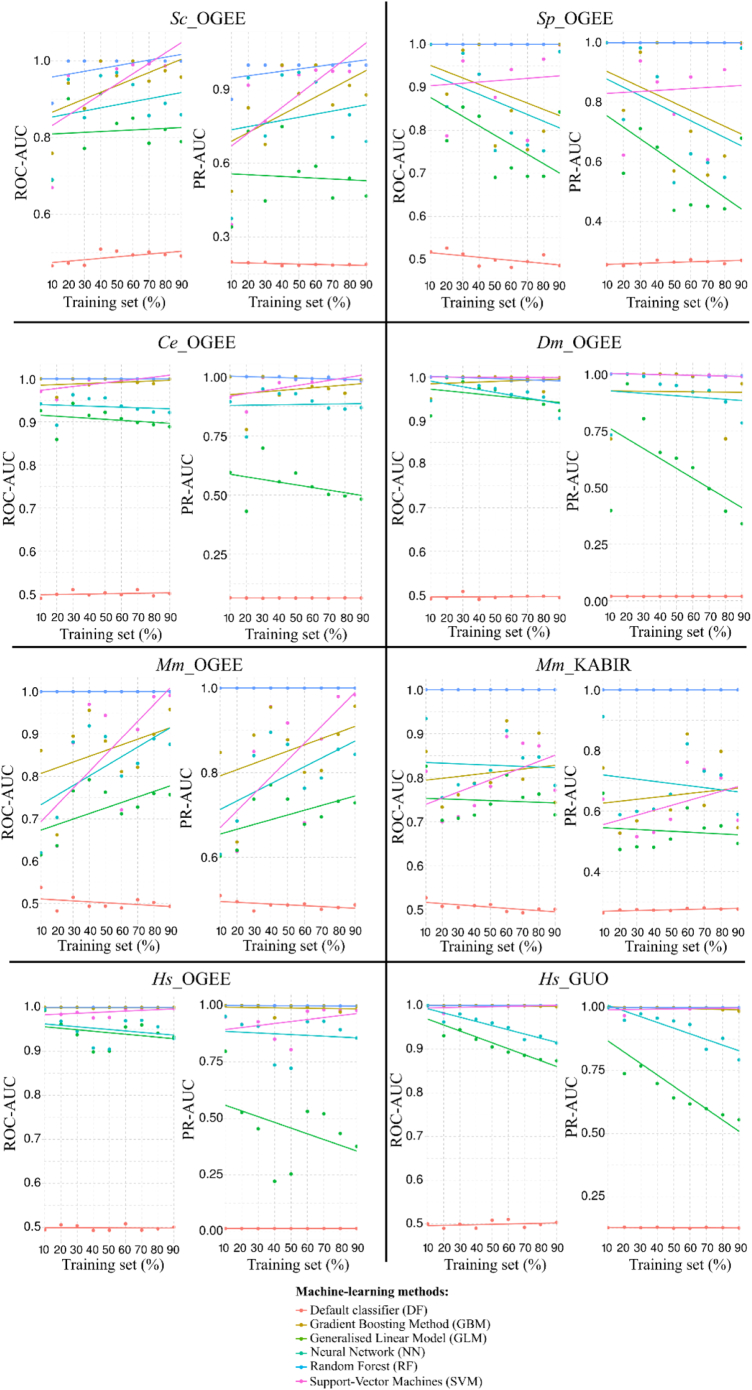
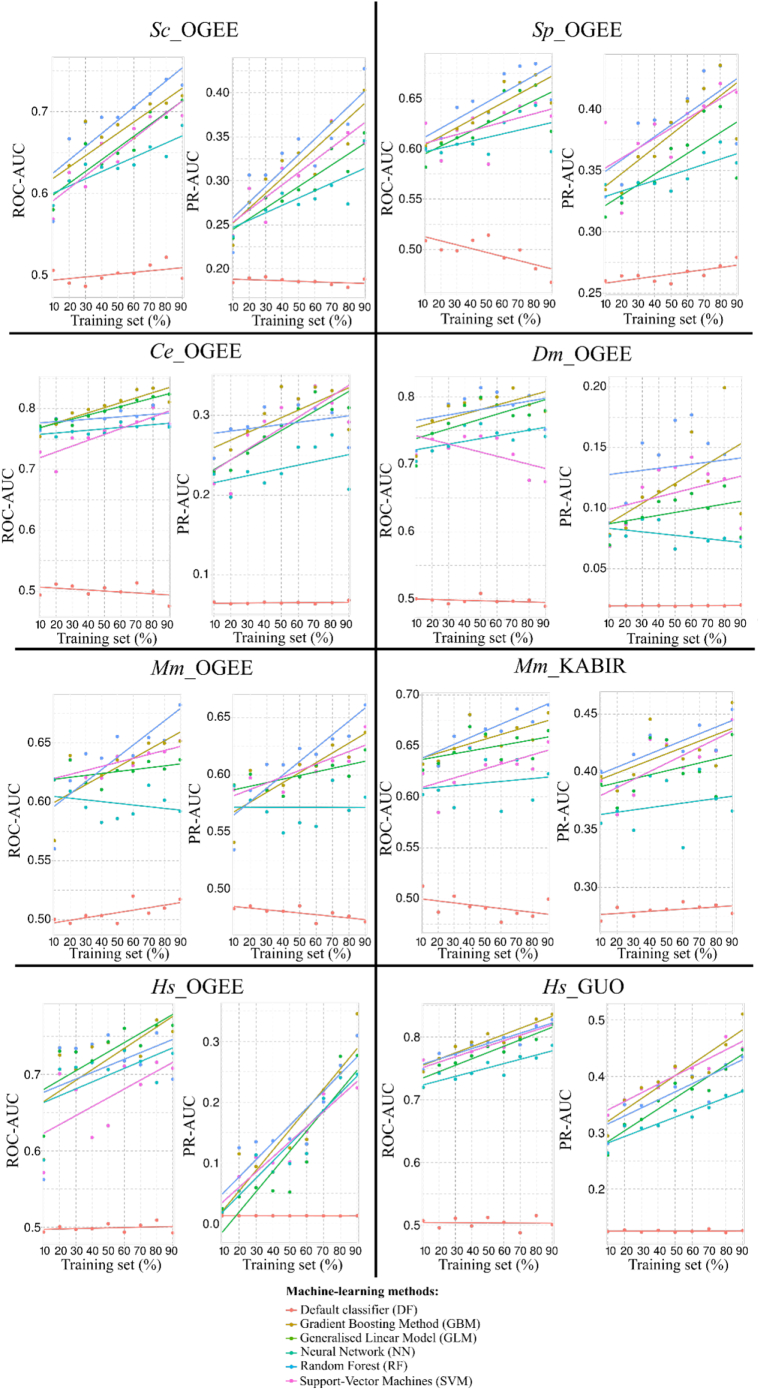
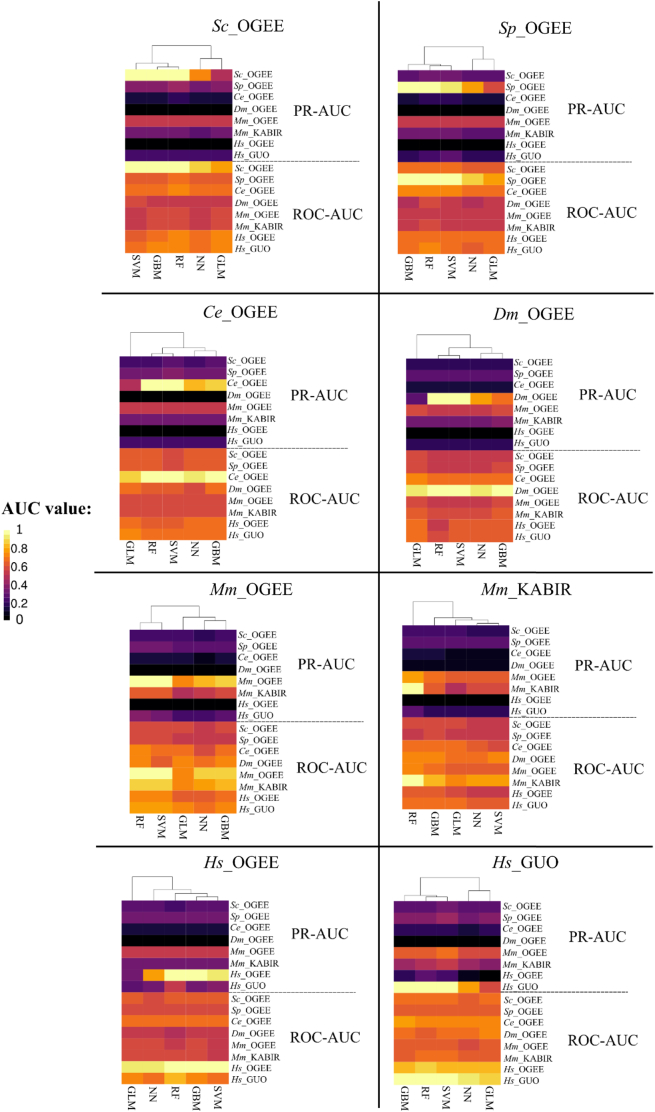
The availability of whole-genome sequences and associated multi-omics data sets, combined with advances in gene knockout and knockdown methods, has enabled large-scale annotation and exploration of gene and protein functions in eukaryotes. Knowing which genes are essential for the survival of eukaryotic organisms is paramount for an understanding of the basic mechanisms of life, and could assist in identifying intervention targets in eukaryotic pathogens and cancer. Here, we studied essential gene orthologs among selected species of eukaryotes, and then employed a systematic machine-learning approach, using protein sequence-derived features and selection procedures, to investigate essential gene predictions within and among species. We showed that the numbers of essential gene orthologs comprise small fractions when compared with the total number of orthologs among the eukaryotic species studied. In addition, we demonstrated that machine-learning models trained with subsets of essentiality-related data performed better than random guessing of gene essentiality for a particular species. Consistent with our gene ortholog analysis, the predictions of essential genes among multiple (including distantly-related) species is possible, yet challenging, suggesting that most essential genes are unique to a species. The present work provides a foundation for the expansion of genome-wide essentiality investigations in eukaryotes using machine learning approaches.
全基因组序列及相关多组学数据集的可得性,再加上基因敲除和敲低方法的进展,使得对真核生物中基因和蛋白质功能进行大规模注释和探索成为可能。了解哪些基因对于真核生物的生存至关重要,对于理解生命的基本机制至关重要,并且有助于确定真核病原体和癌症中的干预靶点。在此,我们研究了选定真核生物物种中的必需基因直系同源物,然后采用系统的机器学习方法,利用蛋白质序列衍生特征和选择程序,来研究物种内部和物种之间的必需基因预测。我们表明,与所研究的真核生物物种中的直系同源物总数相比,必需基因直系同源物的数量只占很小的比例。此外,我们证明,用与必需性相关的数据子集训练的机器学习模型,在预测特定物种的基因必需性方面比随机猜测表现更好。与我们的基因直系同源物分析一致,对多个(包括远缘相关的)物种中的必需基因进行预测是可能的,但具有挑战性,这表明大多数必需基因是物种特有的。本研究为使用机器学习方法扩展真核生物全基因组必需性研究奠定了基础。