Sedlazeck Fritz J, Dhroso Andi, Bodian Dale L, Paschall Justin, Hermes Farrah, Zook Justin M
Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX, USA.
Worcester Polytechnic Institute, Worcester, MA, USA.
F1000Res. 2017 Oct 3;6:1795. doi: 10.12688/f1000research.12516.1. eCollection 2017.
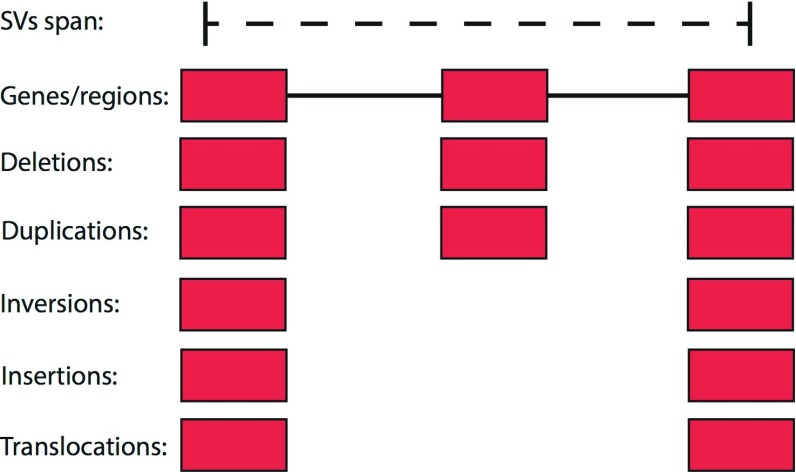
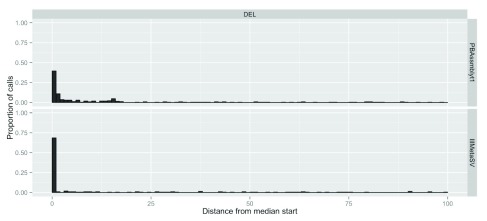
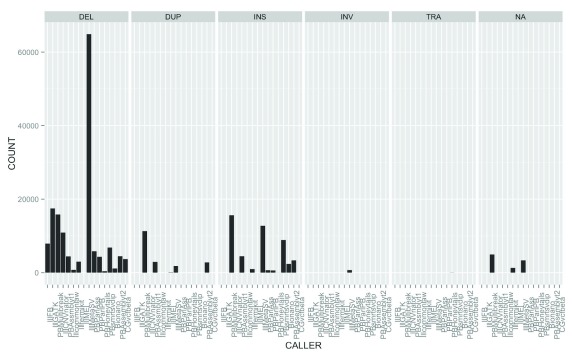
The impact of structural variants (SVs) on a variety of organisms and diseases like cancer has become increasingly evident. Methods for SV detection when studying genomic differences across cells, individuals or populations are being actively developed. Currently, just a few methods are available to compare different SVs callsets, and no specialized methods are available to annotate SVs that account for the unique characteristics of these variant types. Here, we introduce SURVIVOR_ant, a tool that compares types and breakpoints for candidate SVs from different callsets and enables fast comparison of SVs to genomic features such as genes and repetitive regions, as well as to previously established SV datasets such as from the 1000 Genomes Project. As proof of concept we compared 16 SV callsets generated by different SV calling methods on a single genome, the Genome in a Bottle sample HG002 (Ashkenazi son), and annotated the SVs with gene annotations, 1000 Genomes Project SV calls, and four different types of repetitive regions. Computation time to annotate 134,528 SVs with 33,954 of annotations was 22 seconds on a laptop.
结构变异(SVs)对各种生物体及癌症等疾病的影响已日益明显。在研究细胞、个体或群体间的基因组差异时,用于检测SVs的方法正在积极研发中。目前,仅有少数方法可用于比较不同的SVs调用集,且尚无专门方法可注释考虑到这些变异类型独特特征的SVs。在此,我们介绍SURVIVOR_ant,这是一种工具,可比较来自不同调用集的候选SVs的类型和断点,并能快速将SVs与基因和重复区域等基因组特征以及先前建立的SV数据集(如来自千人基因组计划的数据集)进行比较。作为概念验证,我们在单个基因组(瓶中基因组样本HG002,一名德系犹太人男性)上比较了由不同SV检测方法生成的16个SV调用集,并用基因注释、千人基因组计划SV调用以及四种不同类型的重复区域对SVs进行注释。在一台笔记本电脑上,用33,954条注释注释134,528个SVs的计算时间为22秒。