Coudrat Thomas, Simms John, Christopoulos Arthur, Wootten Denise, Sexton Patrick M
Drug Discovery Biology and Department of Pharmacology, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, Victoria, Australia.
School of Life and Health Sciences, Aston University, Birmingham, United Kingdom.
PLoS Comput Biol. 2017 Nov 13;13(11):e1005819. doi: 10.1371/journal.pcbi.1005819. eCollection 2017 Nov.
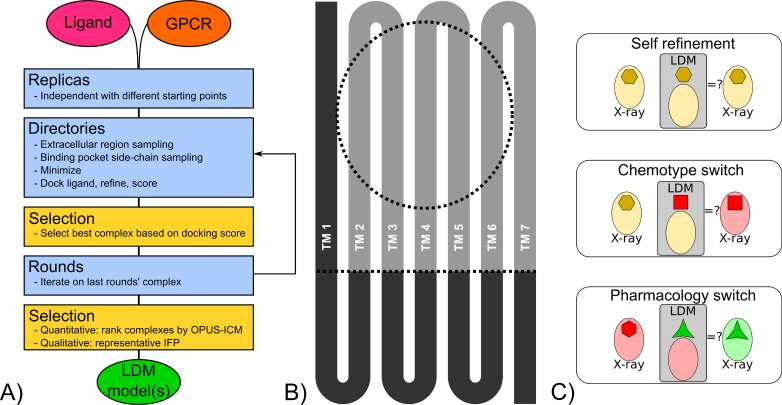
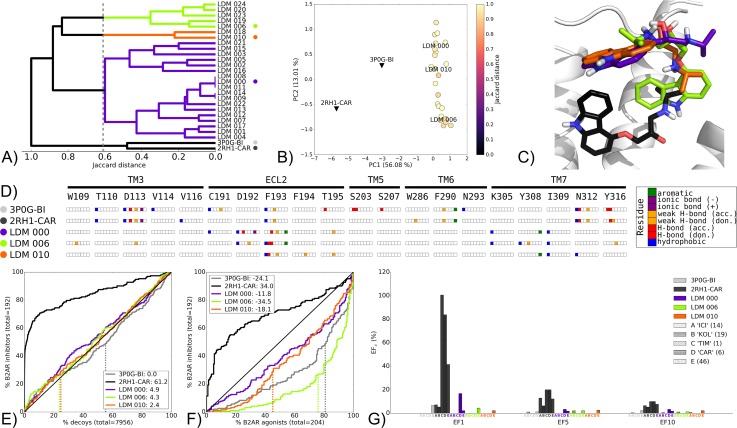
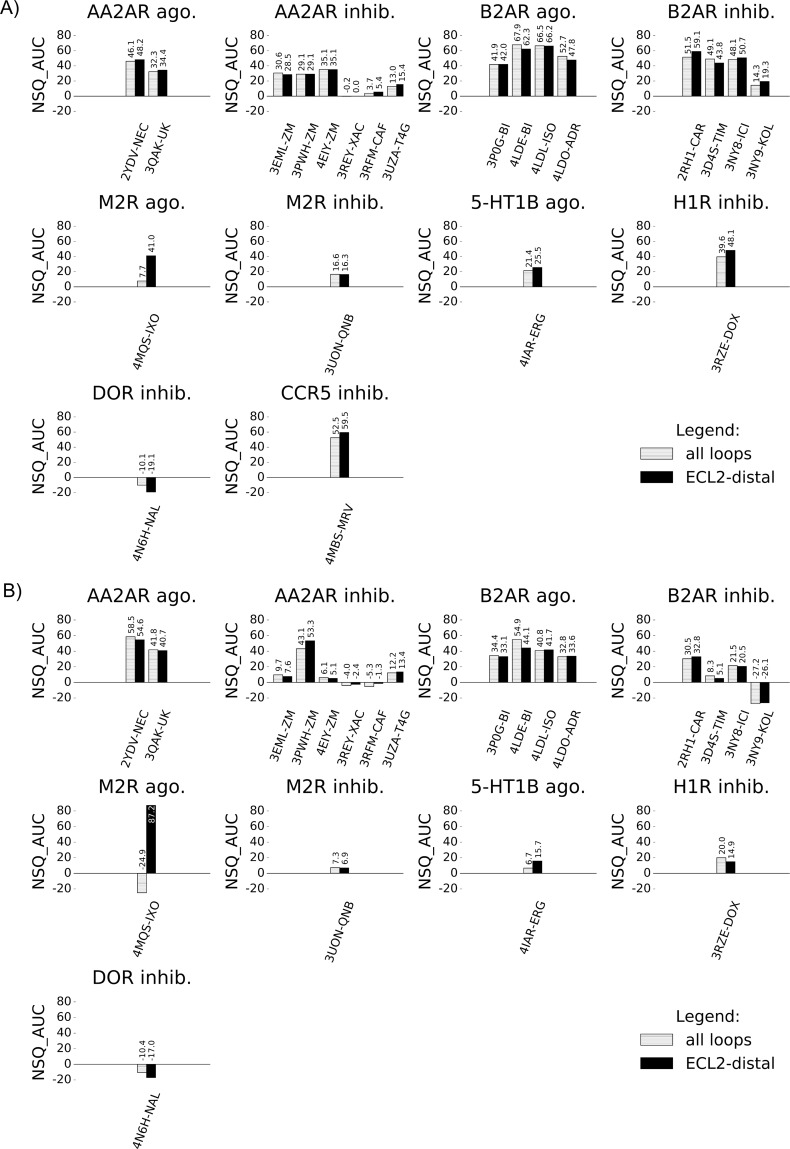
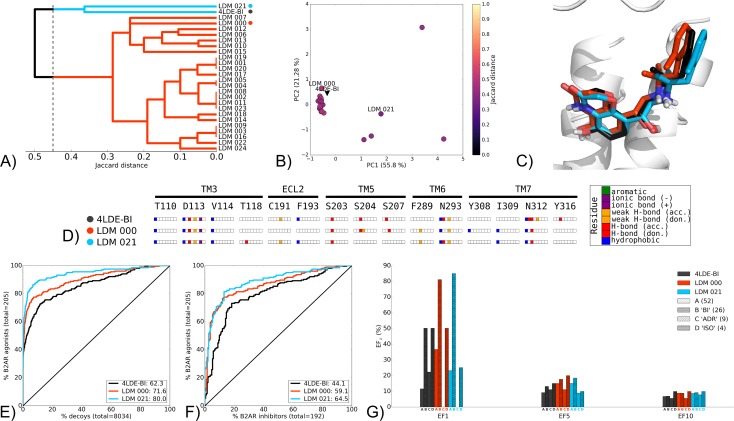
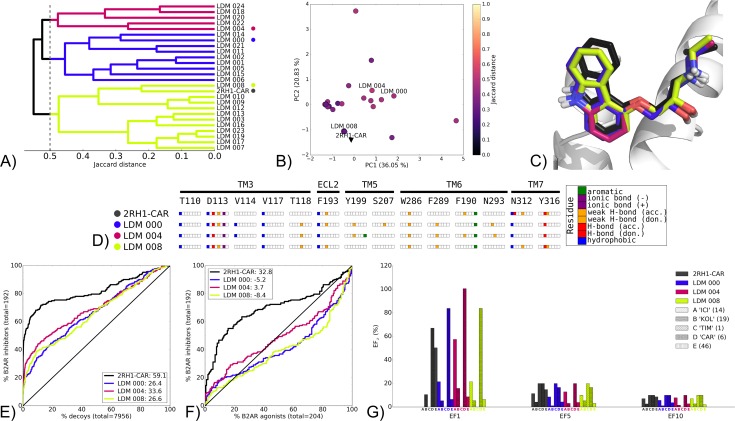
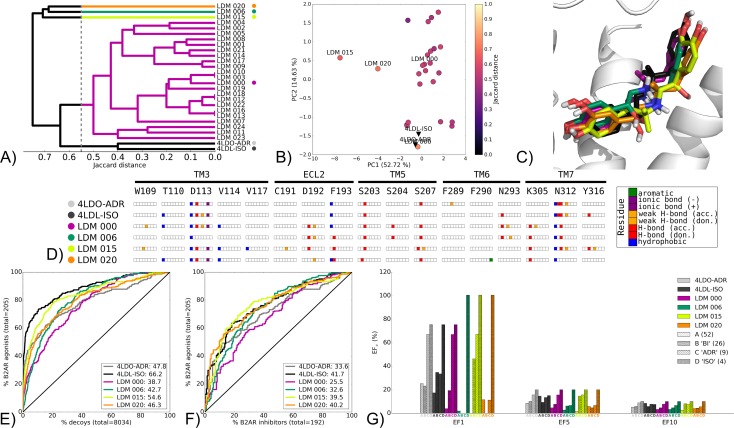
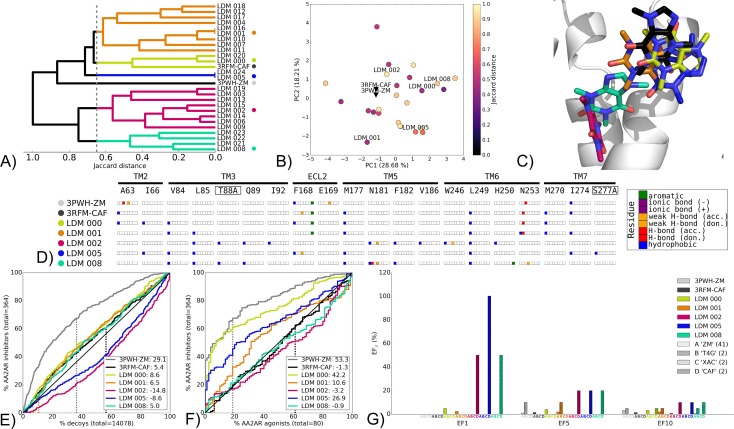
G protein-coupled receptors (GPCRs) play crucial roles in cell physiology and pathophysiology. There is increasing interest in using structural information for virtual screening (VS) of libraries and for structure-based drug design to identify novel agonist or antagonist leads. However, the sparse availability of experimentally determined GPCR/ligand complex structures with diverse ligands impedes the application of structure-based drug design (SBDD) programs directed to identifying new molecules with a select pharmacology. In this study, we apply ligand-directed modeling (LDM) to available GPCR X-ray structures to improve VS performance and selectivity towards molecules of specific pharmacological profile. The described method refines a GPCR binding pocket conformation using a single known ligand for that GPCR. The LDM method is a computationally efficient, iterative workflow consisting of protein sampling and ligand docking. We developed an extensive benchmark comparing LDM-refined binding pockets to GPCR X-ray crystal structures across seven different GPCRs bound to a range of ligands of different chemotypes and pharmacological profiles. LDM-refined models showed improvement in VS performance over origin X-ray crystal structures in 21 out of 24 cases. In all cases, the LDM-refined models had superior performance in enriching for the chemotype of the refinement ligand. This likely contributes to the LDM success in all cases of inhibitor-bound to agonist-bound binding pocket refinement, a key task for GPCR SBDD programs. Indeed, agonist ligands are required for a plethora of GPCRs for therapeutic intervention, however GPCR X-ray structures are mostly restricted to their inactive inhibitor-bound state.
G蛋白偶联受体(GPCRs)在细胞生理和病理生理过程中发挥着关键作用。利用结构信息进行文库虚拟筛选(VS)以及基于结构的药物设计以识别新型激动剂或拮抗剂先导物的兴趣与日俱增。然而,实验测定的具有多种配体的GPCR/配体复合物结构稀少,这阻碍了旨在识别具有特定药理学特性的新分子的基于结构的药物设计(SBDD)程序的应用。在本研究中,我们将配体导向建模(LDM)应用于现有的GPCR X射线结构,以提高VS性能以及对具有特定药理学特征分子的选择性。所描述的方法使用针对该GPCR的单个已知配体来优化GPCR结合口袋构象。LDM方法是一种计算效率高的迭代工作流程,由蛋白质采样和配体对接组成。我们开展了一项广泛的基准测试,将LDM优化的结合口袋与七种不同GPCR的X射线晶体结构进行比较,这些GPCR与一系列不同化学类型和药理学特征的配体结合。在24个案例中的21个案例中,LDM优化的模型在VS性能方面相较于原始X射线晶体结构有所改善。在所有案例中,LDM优化的模型在富集优化配体的化学类型方面具有卓越性能。这可能是LDM在抑制剂结合到激动剂结合口袋优化的所有案例中取得成功的原因,这是GPCR SBDD程序的一项关键任务。事实上,众多GPCR进行治疗干预都需要激动剂配体,然而GPCR X射线结构大多局限于其无活性的抑制剂结合状态。