Computational Biophysics Laboratory (GRIB-IMIM), Universitat Pompeu Fabra, Barcelona Biomedical Research Park (PRBB), Doctor Aiguader 88, 08003, Barcelona, Spain.
Acellera, PRBB, Doctor Aiguader 88, 08003, Barcelona, Spain.
Sci Rep. 2018 Jan 17;8(1):897. doi: 10.1038/s41598-018-19345-7.
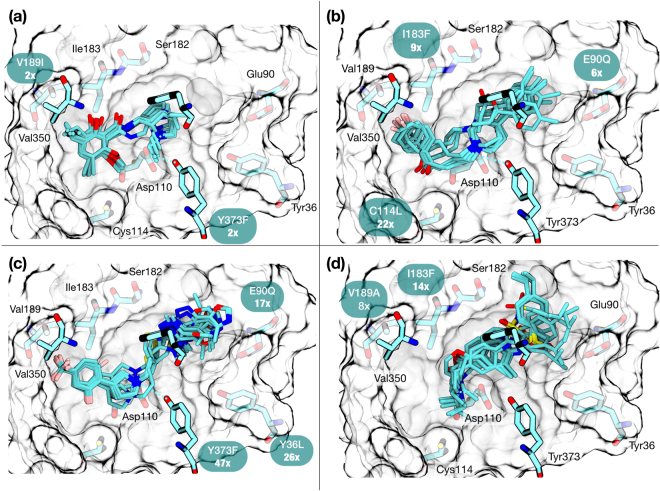
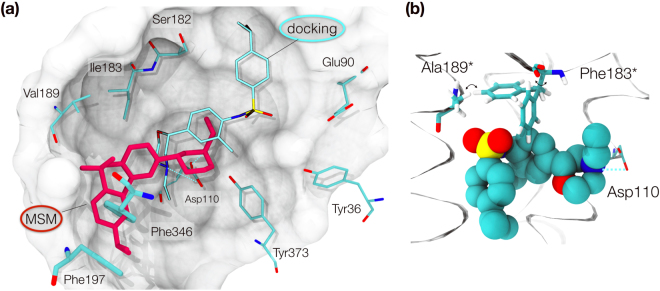
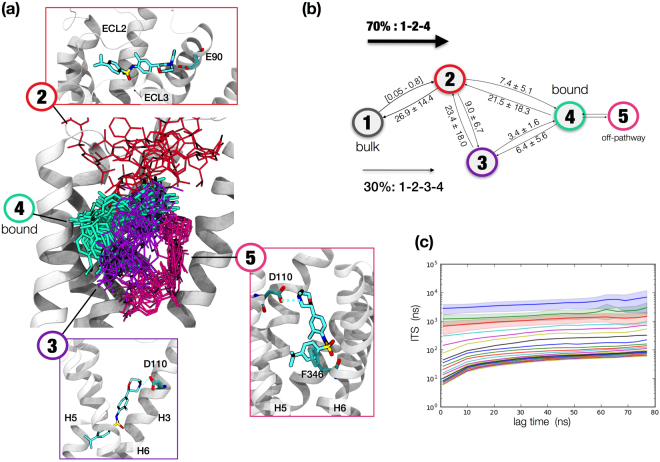
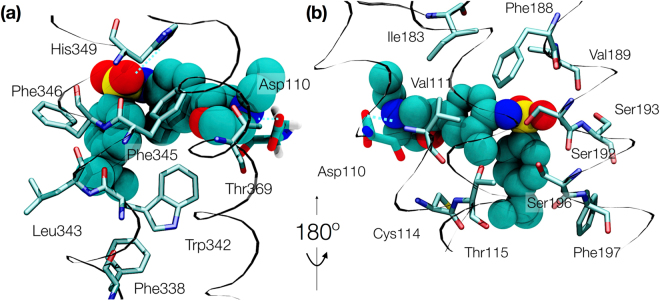
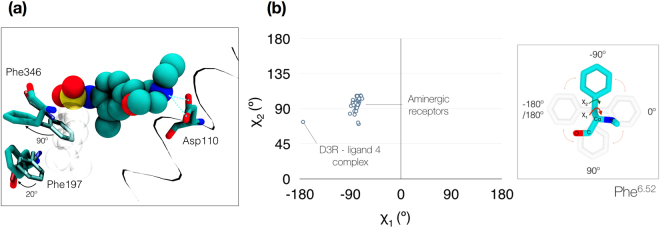
The recent increase in the number of X-ray crystal structures of G-protein coupled receptors (GPCRs) has been enabling for structure-based drug design (SBDD) efforts. These structures have revealed that GPCRs are highly dynamic macromolecules whose function is dependent on their intrinsic flexibility. Unfortunately, the use of static structures to understand ligand binding can potentially be misleading, especially in systems with an inherently high degree of conformational flexibility. Here, we show that docking a set of dopamine D3 receptor compounds into the existing eticlopride-bound dopamine D3 receptor (D3R) X-ray crystal structure resulted in poses that were not consistent with results obtained from site-directed mutagenesis experiments. We overcame the limitations of static docking by using large-scale high-throughput molecular dynamics (MD) simulations and Markov state models (MSMs) to determine an alternative pose consistent with the mutation data. The new pose maintains critical interactions observed in the D3R/eticlopride X-ray crystal structure and suggests that a cryptic pocket forms due to the shift of a highly conserved residue, F. Our study highlights the importance of GPCR dynamics to understand ligand binding and provides new opportunities for drug discovery.
近年来,G 蛋白偶联受体(GPCR)的 X 射线晶体结构数量不断增加,这使得基于结构的药物设计(SBDD)成为可能。这些结构表明,GPCR 是高度动态的大分子,其功能依赖于其固有灵活性。不幸的是,使用静态结构来理解配体结合可能会产生误导,特别是在固有构象灵活性较高的系统中。在这里,我们表明,将一组多巴胺 D3 受体化合物对接入现有的依替唑仑结合的多巴胺 D3 受体(D3R)X 射线晶体结构中,得到的构象与通过定点突变实验获得的结果不一致。我们通过使用大规模高通量分子动力学(MD)模拟和马尔可夫状态模型(MSM)来克服静态对接的局限性,确定了与突变数据一致的替代构象。新构象保持了在 D3R/依替唑仑 X 射线晶体结构中观察到的关键相互作用,并表明由于高度保守的残基 F 的移位,形成了一个隐匿口袋。我们的研究强调了 GPCR 动力学对理解配体结合的重要性,并为药物发现提供了新的机会。