Sheynkman Gloria M, Johnson James E, Jagtap Pratik D, Shortreed Michael R, Onsongo Getiria, Frey Brian L, Griffin Timothy J, Smith Lloyd M
Chemistry Department, University of Wisconsin-Madison, 1101 University Ave,, Madison, WI 53706, USA.
BMC Genomics. 2014 Aug 22;15(1):703. doi: 10.1186/1471-2164-15-703.
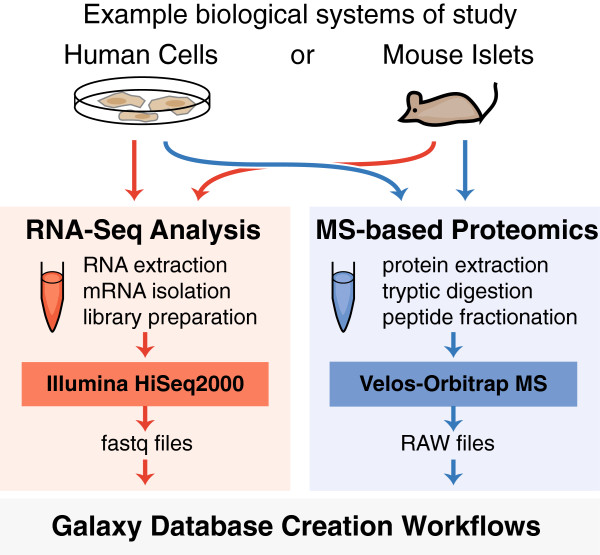
Current practice in mass spectrometry (MS)-based proteomics is to identify peptides by comparison of experimental mass spectra with theoretical mass spectra derived from a reference protein database; however, this strategy necessarily fails to detect peptide and protein sequences that are absent from the database. We and others have recently shown that customized proteomic databases derived from RNA-Seq data can be employed for MS-searching to both improve MS analysis and identify novel peptides. While this general strategy constitutes a significant advance for the discovery of novel protein variations, it has not been readily transferable to other laboratories due to the need for many specialized software tools. To address this problem, we have implemented readily accessible, modifiable, and extensible workflows within Galaxy-P, short for Galaxy for Proteomics, a web-based bioinformatic extension of the Galaxy framework for the analysis of multi-omics (e.g. genomics, transcriptomics, proteomics) data.
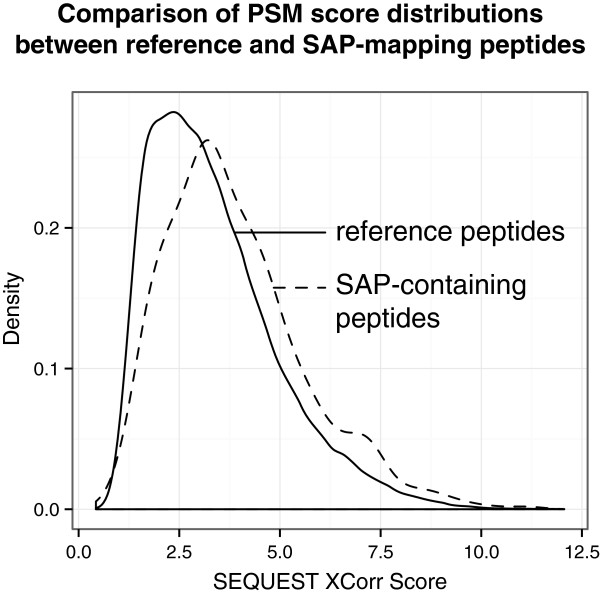
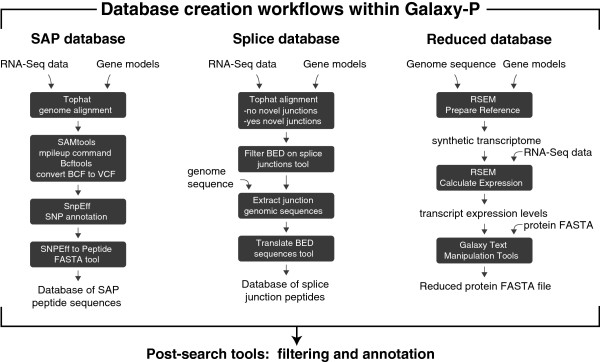
We present three bioinformatic workflows that allow the user to upload raw RNA sequencing reads and convert the data into high-quality customized proteomic databases suitable for MS searching. We show the utility of these workflows on human and mouse samples, identifying 544 peptides containing single amino acid polymorphisms (SAPs) and 187 peptides corresponding to unannotated splice junction peptides, correlating protein and transcript expression levels, and providing the option to incorporate transcript abundance measures within the MS database search process (reduced databases, incorporation of transcript abundance for protein identification score calculations, etc.).
Using RNA-Seq data to enhance MS analysis is a promising strategy to discover novel peptides specific to a sample and, more generally, to improve proteomics results. The main bottleneck for widespread adoption of this strategy has been the lack of easily used and modifiable computational tools. We provide a solution to this problem by introducing a set of workflows within the Galaxy-P framework that converts raw RNA-Seq data into customized proteomic databases.
基于质谱(MS)的蛋白质组学的当前做法是通过将实验质谱与从参考蛋白质数据库衍生的理论质谱进行比较来鉴定肽段;然而,这种策略必然无法检测数据库中不存在的肽段和蛋白质序列。我们和其他人最近表明,源自RNA测序数据的定制蛋白质组学数据库可用于质谱搜索,以改善质谱分析并鉴定新的肽段。虽然这种一般策略在发现新的蛋白质变异方面取得了重大进展,但由于需要许多专门的软件工具,它尚未容易地转移到其他实验室。为了解决这个问题,我们在Galaxy-P(蛋白质组学的Galaxy)中实现了易于访问、可修改和可扩展的工作流程,Galaxy-P是Galaxy框架的基于网络的生物信息学扩展,用于分析多组学(如基因组学、转录组学、蛋白质组学)数据。
我们展示了三种生物信息学工作流程,允许用户上传原始RNA测序读数,并将数据转换为适合质谱搜索的高质量定制蛋白质组学数据库。我们展示了这些工作流程在人类和小鼠样本上的效用,鉴定了544个包含单氨基酸多态性(SAPs)的肽段和187个对应于未注释剪接连接肽段的肽段,关联了蛋白质和转录本表达水平,并提供了在质谱数据库搜索过程中纳入转录本丰度测量的选项(简化数据库、纳入转录本丰度以计算蛋白质鉴定分数等)。
使用RNA测序数据增强质谱分析是发现样本特异性新肽段以及更广泛地改善蛋白质组学结果的有前途的策略。广泛采用该策略的主要瓶颈一直是缺乏易于使用和可修改的计算工具。我们通过在Galaxy-P框架内引入一组将原始RNA测序数据转换为定制蛋白质组学数据库的工作流程来解决这个问题。