Barazandeh Marjan, Lambert Samuel A, Albu Mihai, Hughes Timothy R
Terrence Donnelly Centre for Cellular and Biomolecular Research, University of Toronto, Ontario M5S 1A8, Canada.
Department of Molecular Genetics, University of Toronto, Ontario M5S 1A8, Canada.
G3 (Bethesda). 2018 Jan 4;8(1):219-229. doi: 10.1534/g3.117.300296.
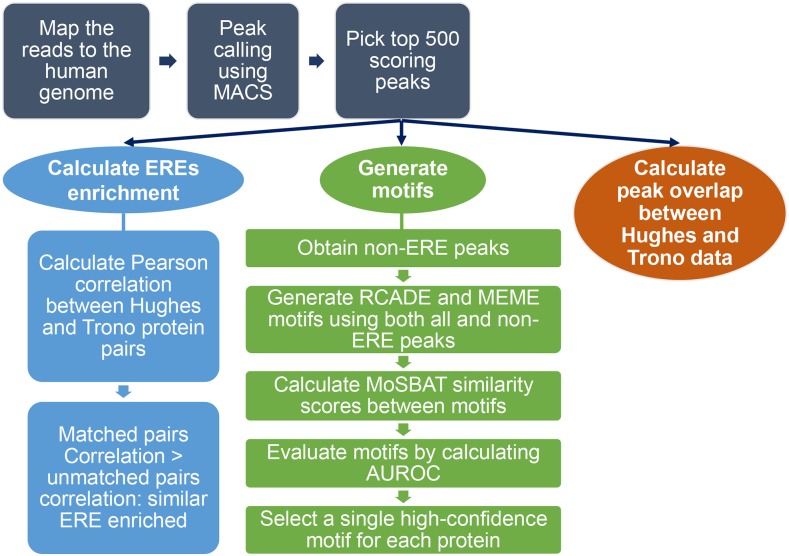
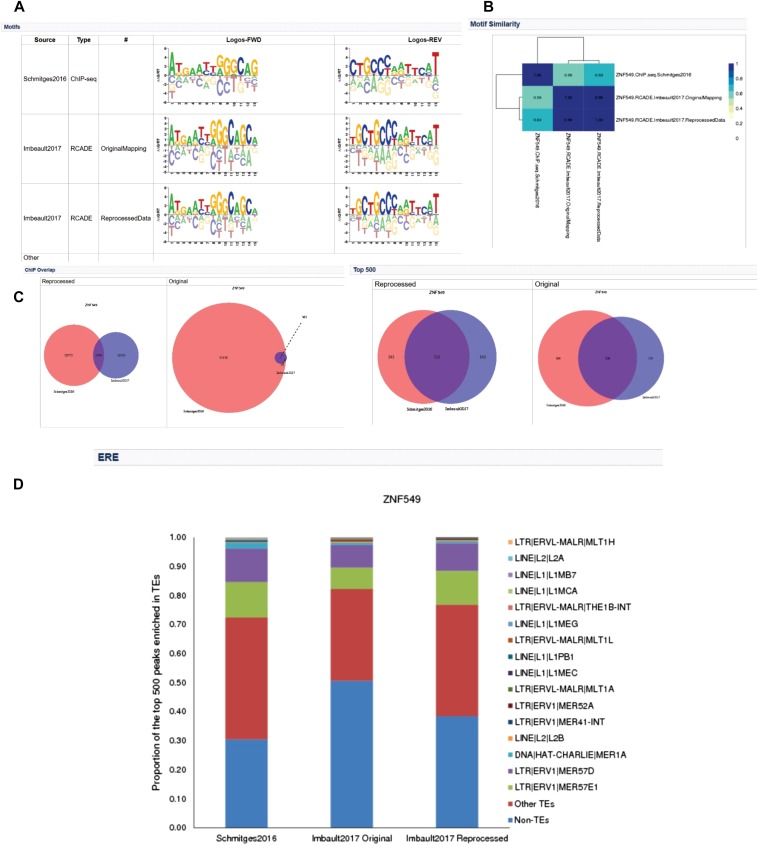
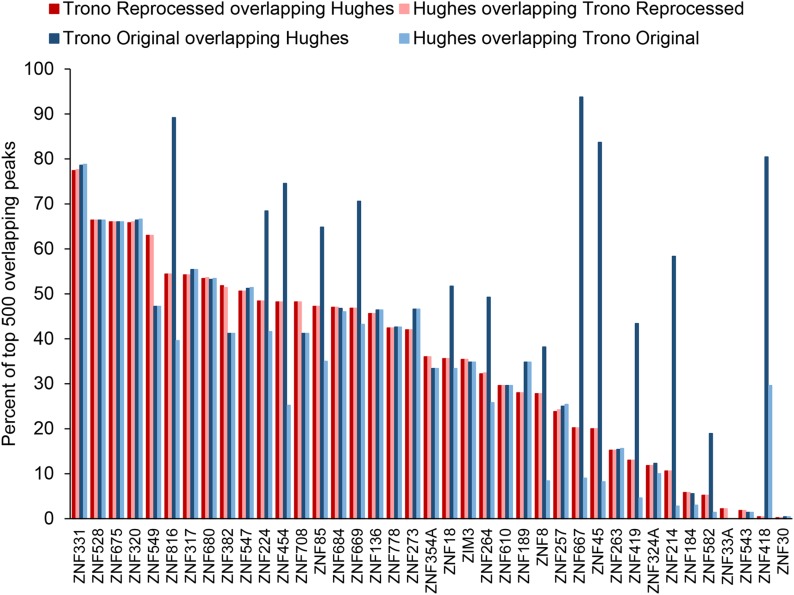
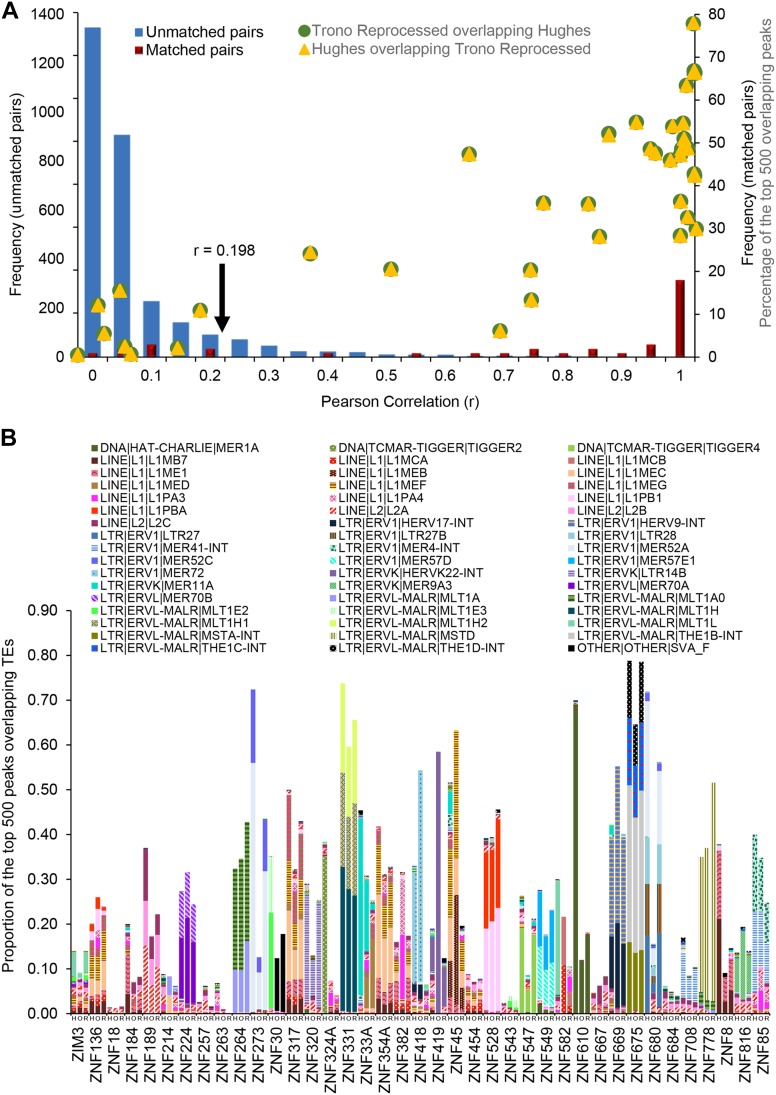
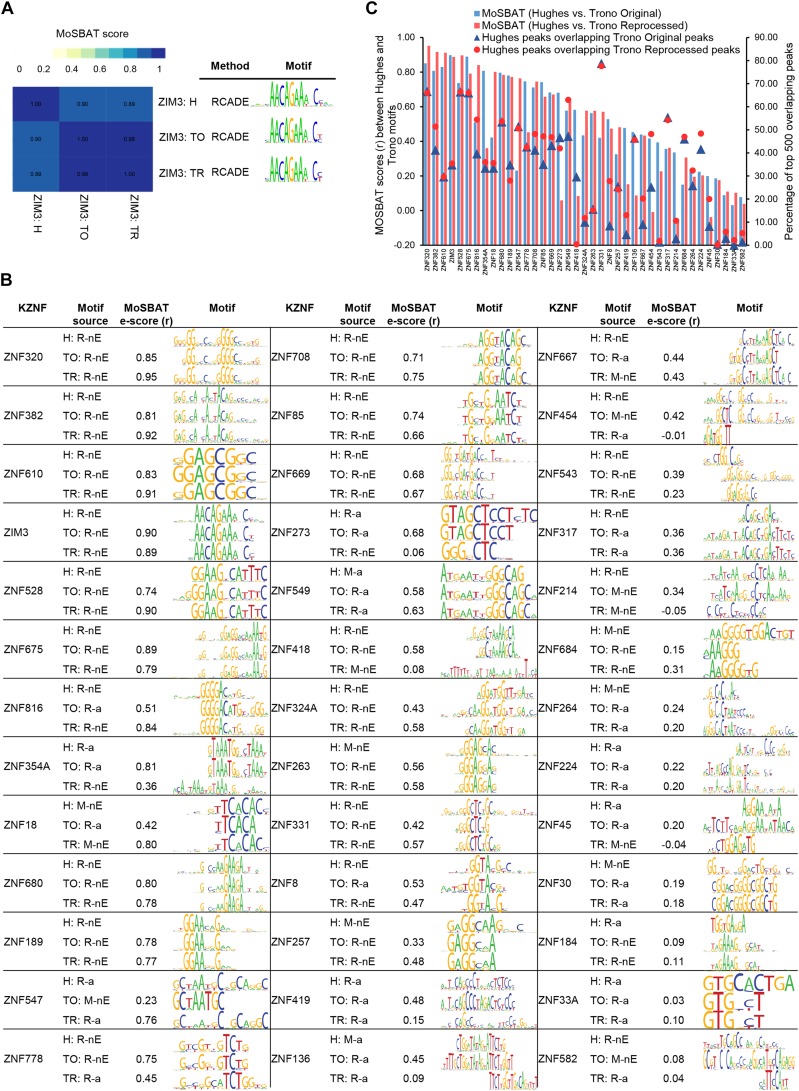
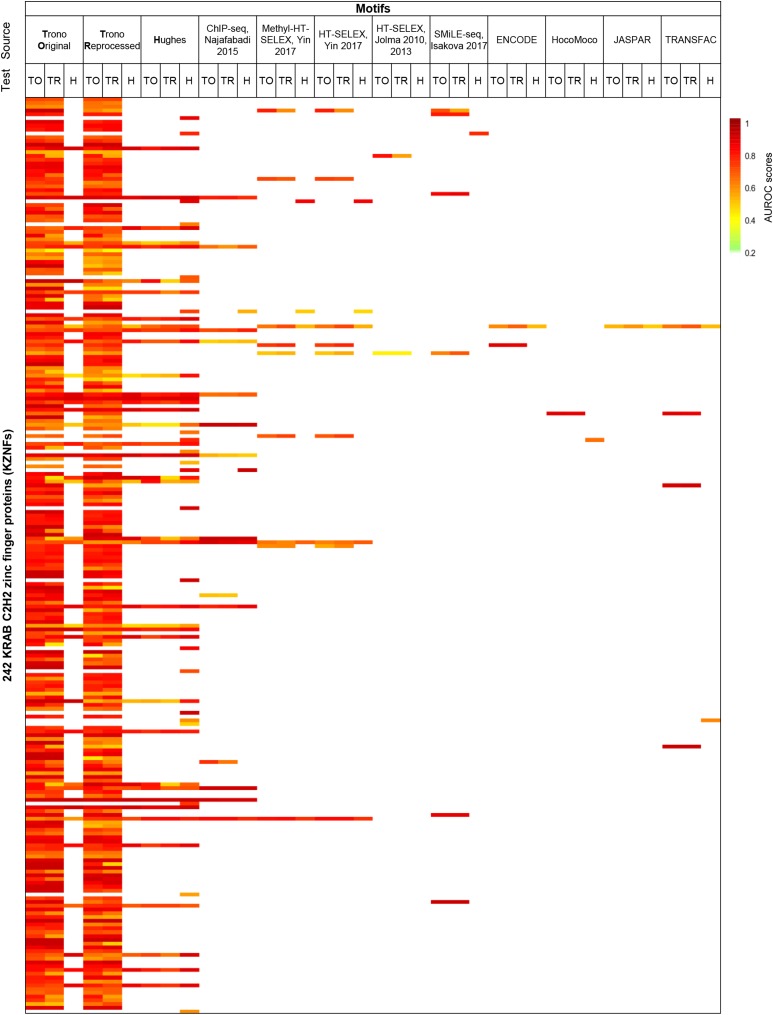
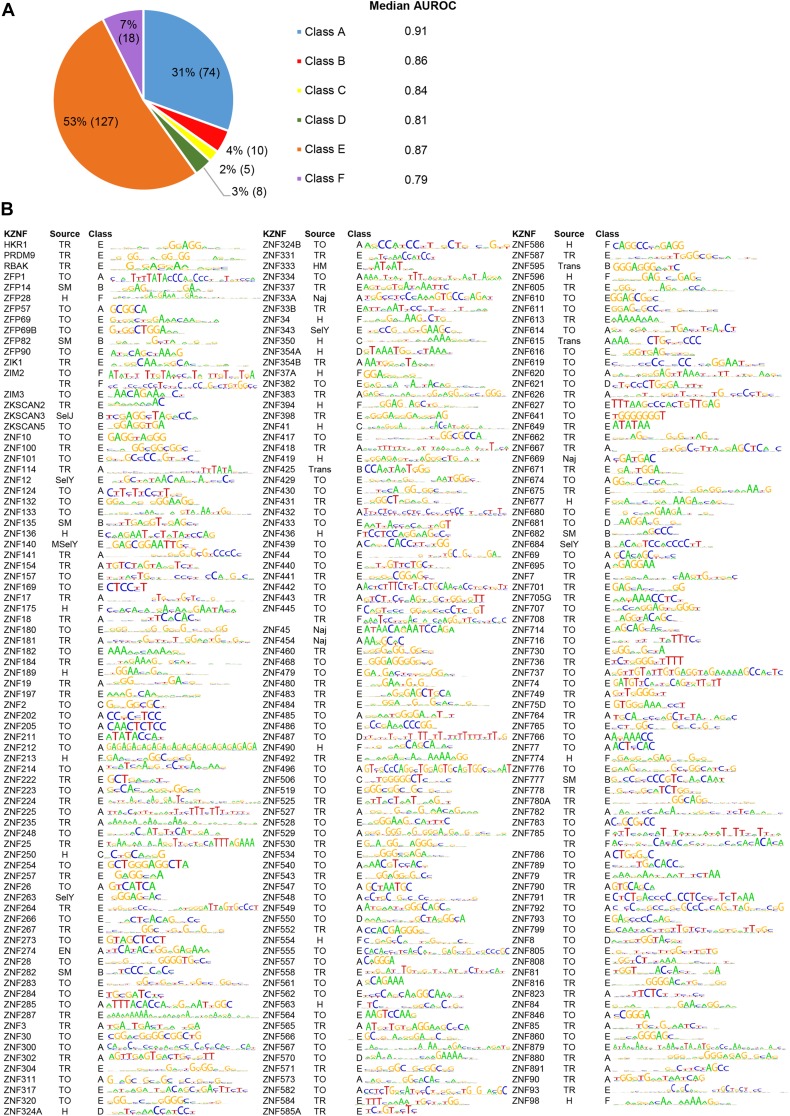
KRAB C2H2 zinc finger proteins (KZNFs) are the largest and most diverse family of human transcription factors, likely due to diversifying selection driven by novel endogenous retroelements (EREs), but the vast majority lack binding motifs or functional data. Two recent studies analyzed a majority of the human KZNFs using either ChIP-seq (60 proteins) or ChIP-exo (221 proteins) in the same cell type (HEK293). The ChIP-exo paper did not describe binding motifs, however. Thirty-nine proteins are represented in both studies, enabling the systematic comparison of the data sets presented here. Typically, only a minority of peaks overlap, but the two studies nonetheless display significant similarity in ERE binding for 32/39, and yield highly similar DNA binding motifs for 23 and related motifs for 34 (MoSBAT similarity score >0.5 and >0.2, respectively). Thus, there is overall (albeit imperfect) agreement between the two studies. For the 242 proteins represented in at least one study, we selected a highest-confidence motif for each protein, utilizing several motif-derivation approaches, and evaluating motifs within and across data sets. Peaks for the majority (158) are enriched (96% with AUC >0.6 predicting peak nonpeak) for a motif that is supported by the C2H2 "recognition code," consistent with intrinsic sequence specificity driving DNA binding in cells. An additional 63 yield motifs enriched in peaks, but not supported by the recognition code, which could reflect indirect binding. Altogether, these analyses validate both data sets, and provide a reference motif set with associated quality metrics.
KRAB C2H2锌指蛋白(KZNFs)是人类转录因子中最大且最多样化的家族,这可能是由于新型内源性逆转录元件(ERE)驱动的多样化选择所致,但绝大多数KZNFs缺乏结合基序或功能数据。最近的两项研究在同一细胞类型(HEK293)中使用ChIP-seq(60种蛋白)或ChIP-exo(221种蛋白)分析了大多数人类KZNFs。然而,ChIP-exo论文并未描述结合基序。两项研究中有39种蛋白是共有的,这使得我们能够对这里呈现的数据集进行系统比较。通常,只有少数峰重叠,但两项研究在32/39的ERE结合方面仍显示出显著相似性,并且为23种蛋白产生了高度相似的DNA结合基序,为34种蛋白产生了相关基序(MoSBAT相似性得分分别>0.5和>0.2)。因此,两项研究总体上(尽管并不完美)达成了一致。对于至少在一项研究中出现的242种蛋白,我们利用多种基序推导方法,为每种蛋白选择了一个最高置信度的基序,并在数据集内部和数据集之间评估基序。大多数(158种)蛋白的峰对于一个由C2H2“识别密码”支持的基序是富集的(96%的曲线下面积>AUC>0.6预测峰与非峰),这与驱动细胞中DNA结合的内在序列特异性一致。另外63种蛋白产生的基序在峰中富集,但不受识别密码支持,这可能反映了间接结合。总之,这些分析验证了两个数据集,并提供了一个带有相关质量指标的参考基序集。