Lind Abigail L, Wisecaver Jennifer H, Lameiras Catarina, Wiemann Philipp, Palmer Jonathan M, Keller Nancy P, Rodrigues Fernando, Goldman Gustavo H, Rokas Antonis
Department of Biomedical Informatics, Vanderbilt University School of Medicine, Nashville, Tennessee, United States of America.
Department of Biological Sciences, Vanderbilt University, Nashville, Tennessee, United States of America.
PLoS Biol. 2017 Nov 17;15(11):e2003583. doi: 10.1371/journal.pbio.2003583. eCollection 2017 Nov.
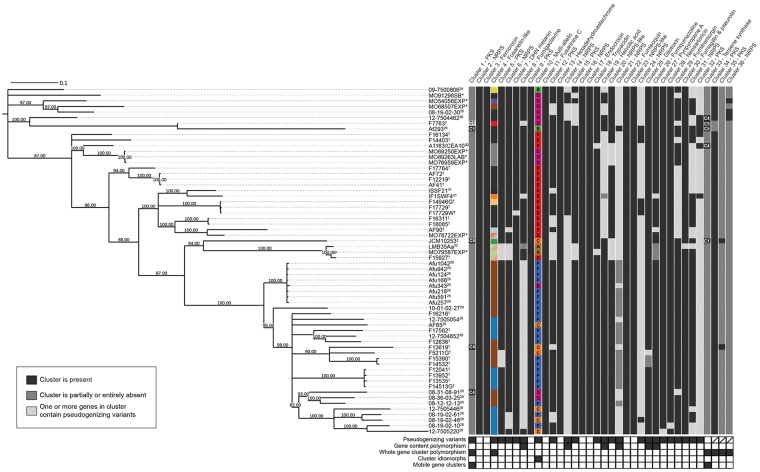
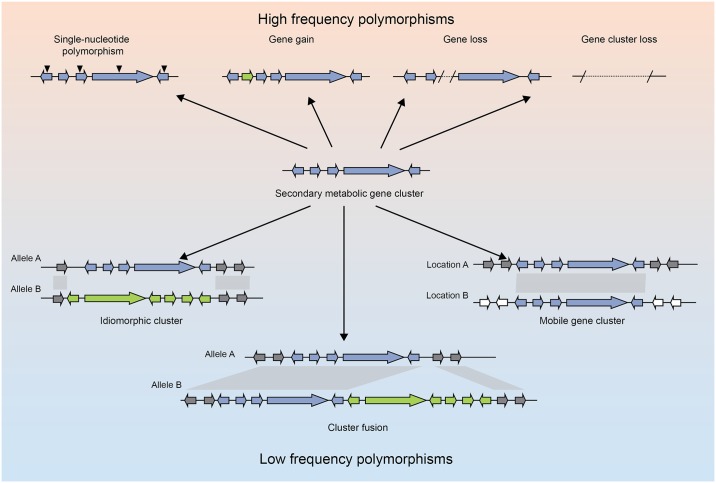
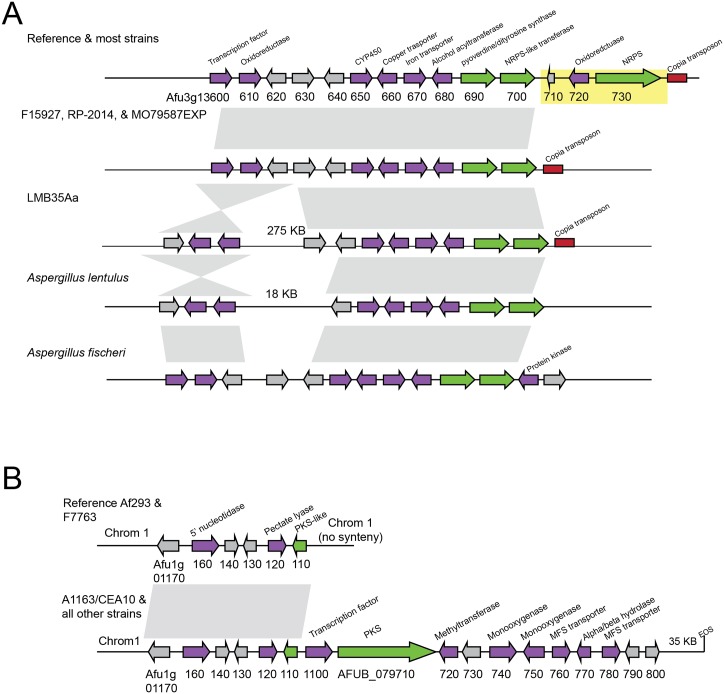
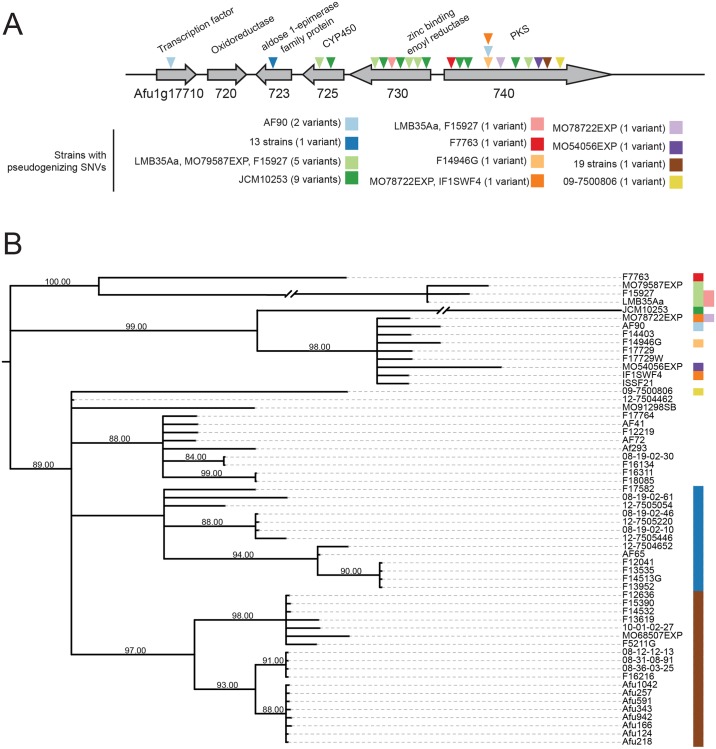
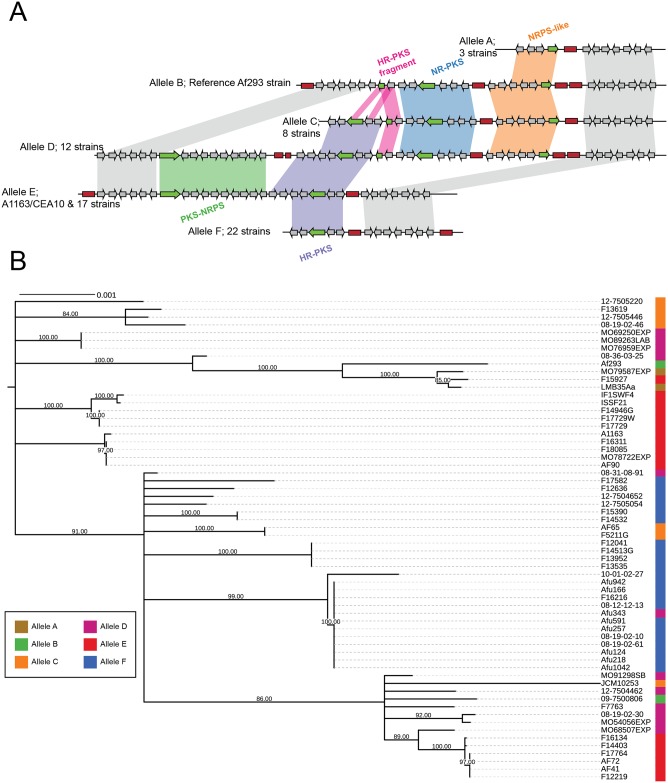
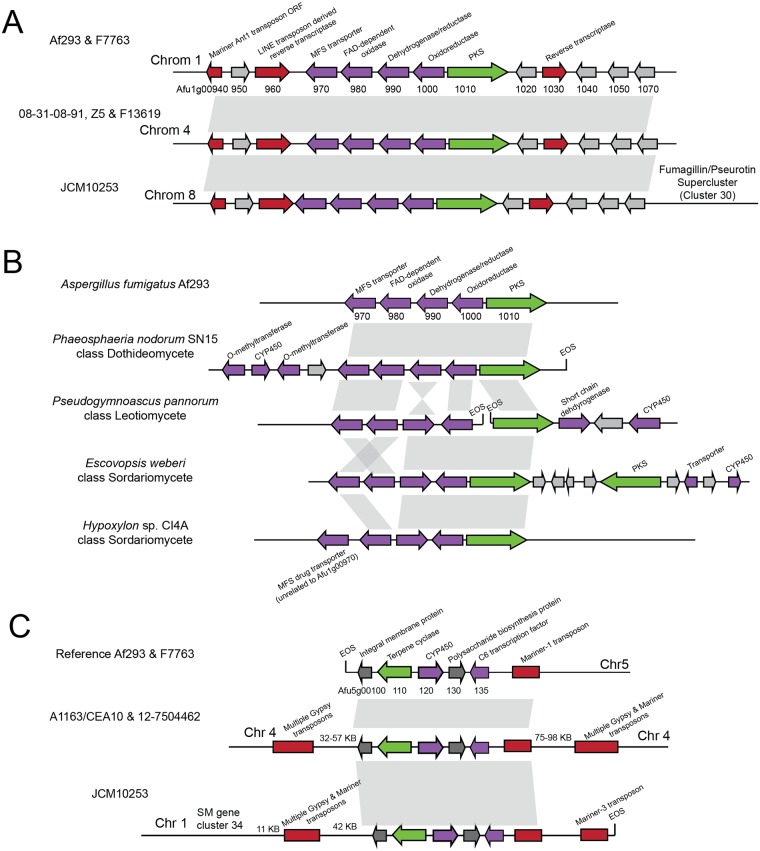
Filamentous fungi produce a diverse array of secondary metabolites (SMs) critical for defense, virulence, and communication. The metabolic pathways that produce SMs are found in contiguous gene clusters in fungal genomes, an atypical arrangement for metabolic pathways in other eukaryotes. Comparative studies of filamentous fungal species have shown that SM gene clusters are often either highly divergent or uniquely present in one or a handful of species, hampering efforts to determine the genetic basis and evolutionary drivers of SM gene cluster divergence. Here, we examined SM variation in 66 cosmopolitan strains of a single species, the opportunistic human pathogen Aspergillus fumigatus. Investigation of genome-wide within-species variation revealed 5 general types of variation in SM gene clusters: nonfunctional gene polymorphisms; gene gain and loss polymorphisms; whole cluster gain and loss polymorphisms; allelic polymorphisms, in which different alleles corresponded to distinct, nonhomologous clusters; and location polymorphisms, in which a cluster was found to differ in its genomic location across strains. These polymorphisms affect the function of representative A. fumigatus SM gene clusters, such as those involved in the production of gliotoxin, fumigaclavine, and helvolic acid as well as the function of clusters with undefined products. In addition to enabling the identification of polymorphisms, the detection of which requires extensive genome-wide synteny conservation (e.g., mobile gene clusters and nonhomologous cluster alleles), our approach also implicated multiple underlying genetic drivers, including point mutations, recombination, and genomic deletion and insertion events as well as horizontal gene transfer from distant fungi. Finally, most of the variants that we uncover within A. fumigatus have been previously hypothesized to contribute to SM gene cluster diversity across entire fungal classes and phyla. We suggest that the drivers of genetic diversity operating within a fungal species shown here are sufficient to explain SM cluster macroevolutionary patterns.
丝状真菌产生多种对防御、毒力和交流至关重要的次级代谢产物(SMs)。产生SMs的代谢途径存在于真菌基因组中相邻的基因簇中,这在其他真核生物的代谢途径中是一种非典型的排列方式。对丝状真菌物种的比较研究表明,SM基因簇通常要么高度分化,要么仅在一个或少数几个物种中独特存在,这阻碍了确定SM基因簇分化的遗传基础和进化驱动因素的努力。在这里,我们研究了单一物种——机会性人类病原体烟曲霉的66个世界性菌株中的SM变异。对全基因组种内变异的研究揭示了SM基因簇的5种一般变异类型:无功能基因多态性;基因获得和缺失多态性;整个簇的获得和缺失多态性;等位基因多态性,其中不同等位基因对应于不同的、非同源的簇;以及位置多态性,其中发现一个簇在不同菌株中的基因组位置不同。这些多态性影响了烟曲霉代表性SM基因簇的功能,例如那些参与产生gliotoxin、烟曲霉黄青霉素和赫沃酸的基因簇以及产物未定义的簇的功能。除了能够识别多态性(其检测需要广泛的全基因组共线性保守,例如移动基因簇和非同源簇等位基因)之外,我们的方法还涉及多种潜在的遗传驱动因素,包括点突变、重组、基因组缺失和插入事件以及来自远缘真菌的水平基因转移。最后,我们在烟曲霉中发现的大多数变异先前已被假设有助于整个真菌类群和门的SM基因簇多样性。我们认为,这里显示的真菌物种内遗传多样性的驱动因素足以解释SM簇的宏观进化模式。