McCafferty Caitlyn L, Sergeev Yuri V
Ophthalmic Genetics and Visual Function Branch, National Eye Institute, National Institutes of Health, Bethesda, Maryland, United States of America.
PLoS One. 2017 Dec 7;12(12):e0189064. doi: 10.1371/journal.pone.0189064. eCollection 2017.
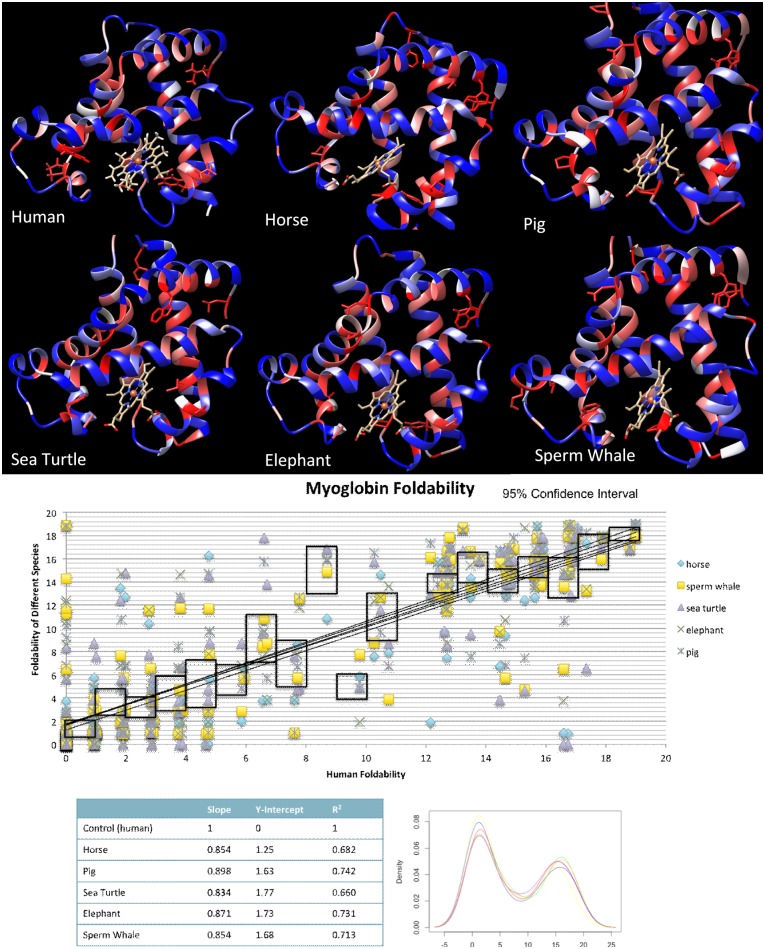
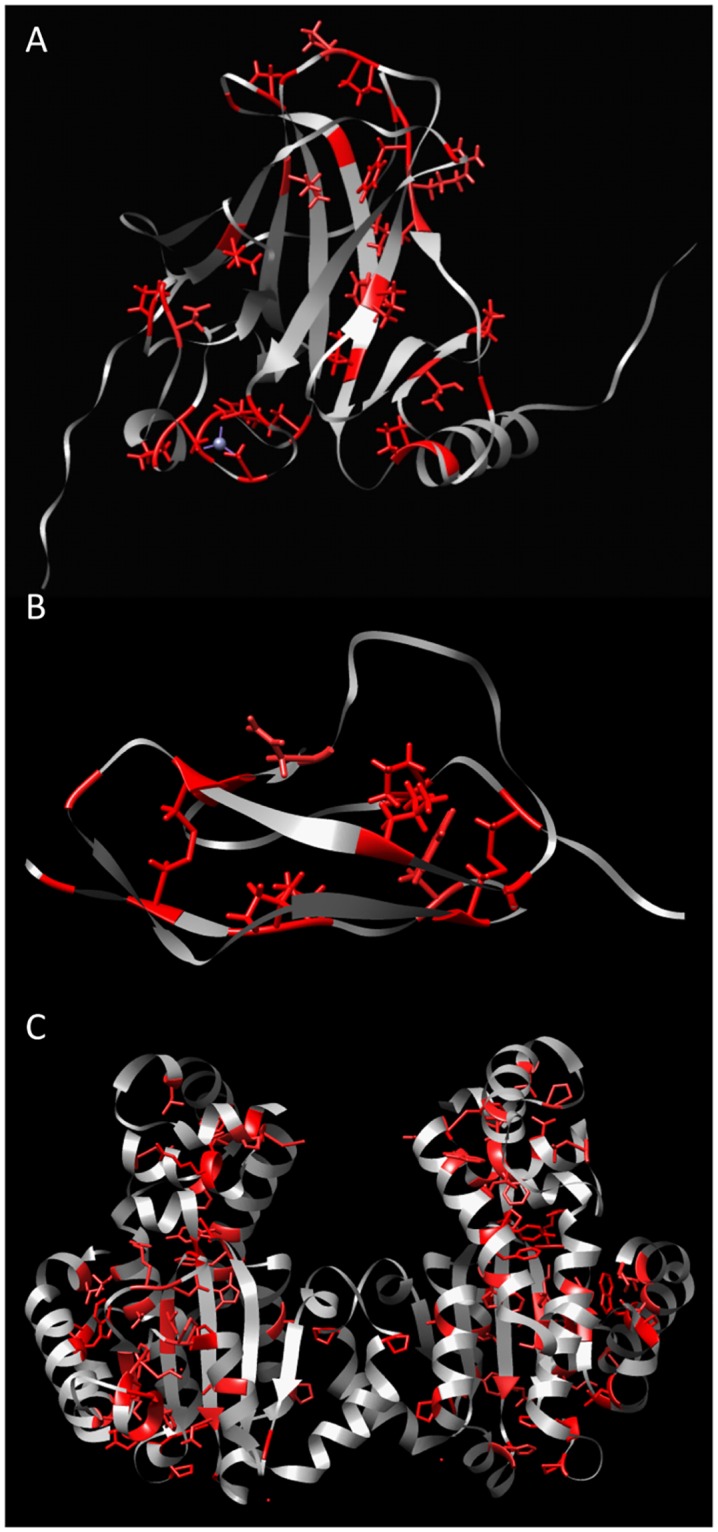
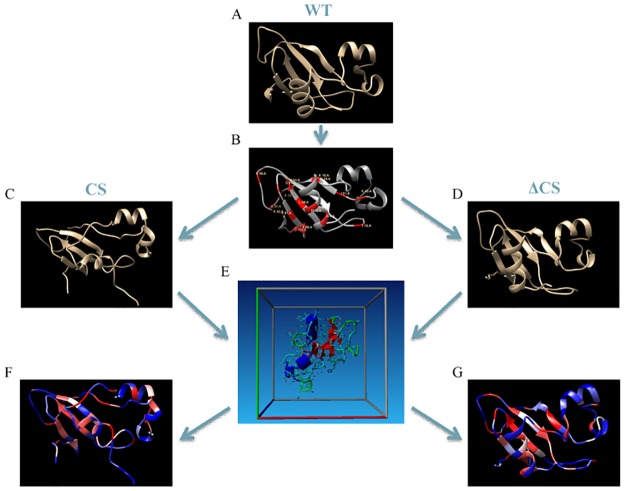
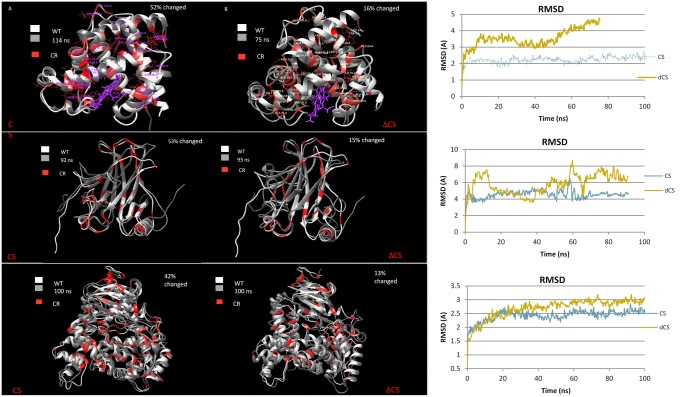
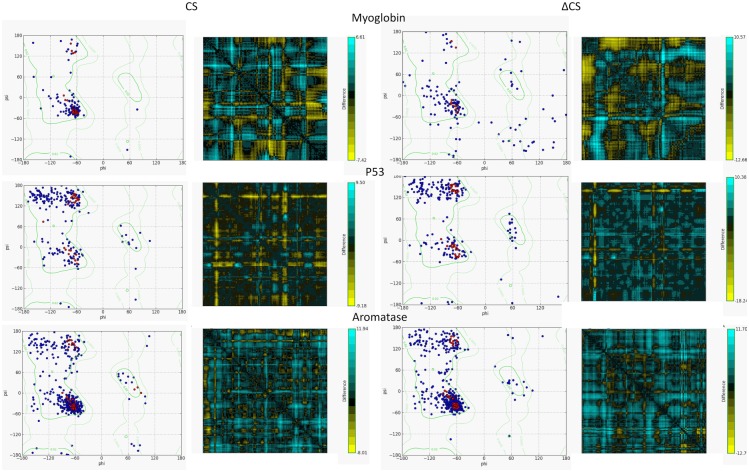
A protein's amino acid sequence dictates the folds and final structure the macromolecule will form. We propose that by identifying critical residues in a protein's atomic structure, we can select a critical stability framework within the protein structure essential to proper protein folding. We use global computational mutagenesis based on the unfolding mutation screen to test the effect of every possible missense mutation on the protein structure to identify the residues that cannot tolerate a substitution without causing protein misfolding. This method was tested using molecular dynamics to simulate the stability effects of mutating critical residues in proteins involved in inherited disease, such as myoglobin, p53, and the 15th sushi domain of complement factor H. In addition we prove that when the critical residues are in place, other residues may be changed within the structure without a stability loss. We validate that critical residues are conserved using myoglobin to show that critical residues are the same for crystal structures of 6 different species and comparing conservation indices to critical residues in 9 eye disease-related proteins. Our studies demonstrate that by using a selection of critical elements in a protein structure we can identify a critical protein stability framework. The frame of critical residues can be used in genetic engineering to improve small molecule binding for drug studies, identify loss-of-function disease-causing missense mutations in genetics studies, and aide in identifying templates for homology modeling.
蛋白质的氨基酸序列决定了该大分子将形成的折叠和最终结构。我们提出,通过识别蛋白质原子结构中的关键残基,我们可以在蛋白质结构中选择一个对蛋白质正确折叠至关重要的关键稳定性框架。我们使用基于展开突变筛选的全局计算诱变来测试每个可能的错义突变对蛋白质结构的影响,以识别那些在不导致蛋白质错误折叠的情况下不能耐受替代的残基。使用分子动力学对参与遗传性疾病的蛋白质(如肌红蛋白、p53和补体因子H的第15个寿司结构域)中的关键残基进行突变的稳定性影响进行了模拟,对该方法进行了测试。此外,我们证明,当关键残基就位时,结构中的其他残基可以改变而不会损失稳定性。我们使用肌红蛋白验证关键残基是保守的,以表明6种不同物种的晶体结构中的关键残基是相同的,并将保守指数与9种与眼部疾病相关的蛋白质中的关键残基进行比较。我们的研究表明,通过在蛋白质结构中选择关键元件,我们可以识别关键的蛋白质稳定性框架。关键残基框架可用于基因工程,以改善药物研究中的小分子结合,在遗传学研究中识别功能丧失的致病错义突变,并有助于识别同源建模的模板。