Department of Computer Science, University of Saskatchewan, Saskatoon, Canada.
Department of Mathematics and Statistics, University of Saskatchewan, Saskatoon, Canada.
BMC Genomics. 2017 Nov 17;18(Suppl 9):862. doi: 10.1186/s12864-017-4227-z.
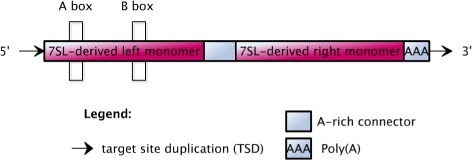
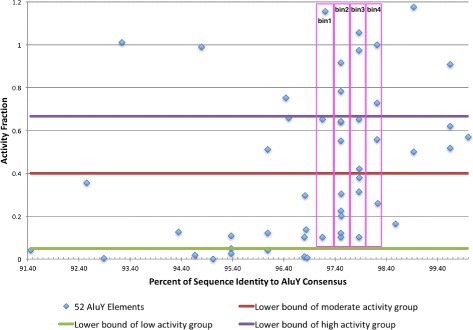
Transposable elements (TEs) are interspersed DNA sequences that can move or copy to new positions within a genome. TEs are believed to promote speciation and their activities play a significant role in human disease. In the human genome, the 22 AluY and 6 AluS TE subfamilies have been the most recently active, and their transposition has been implicated in many inherited human diseases and in various forms of cancer. Therefore, understanding their transposition activity is very important and identifying the factors that affect their transpositional activity is of great interest. Recently, there has been some work done to quantify the activity levels of active Alu TEs based on variation in the sequence. Given this activity data, an analysis of TE activity based on the position of mutations is conducted.
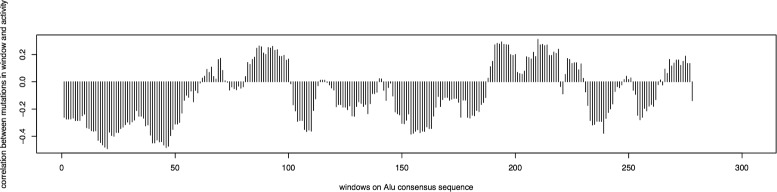
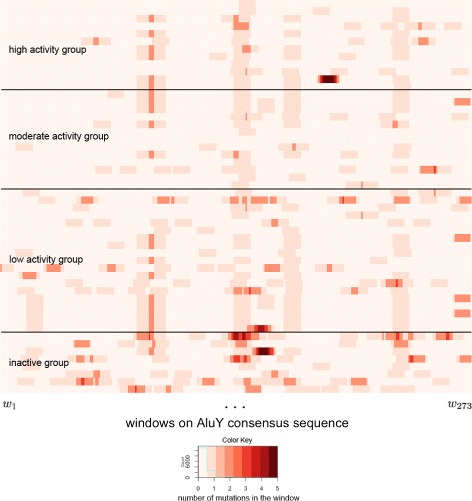
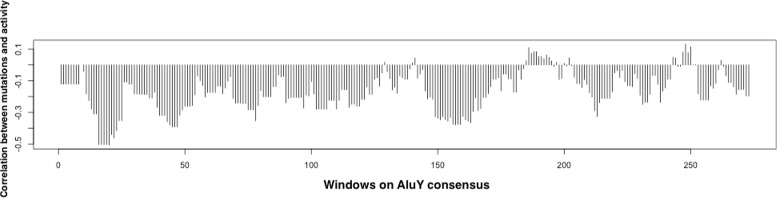
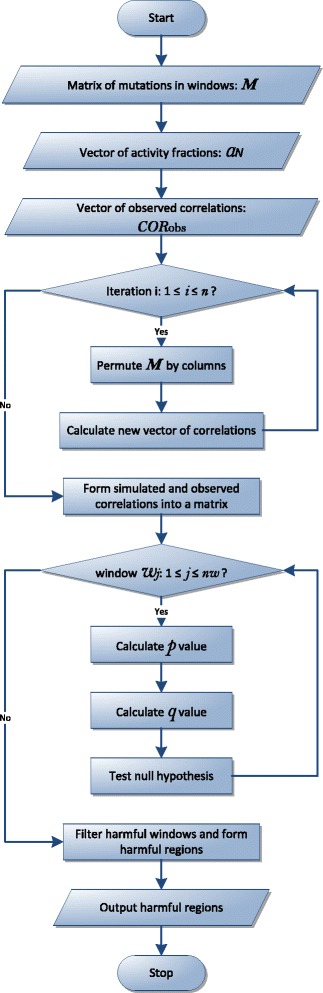
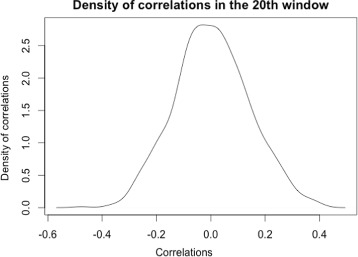
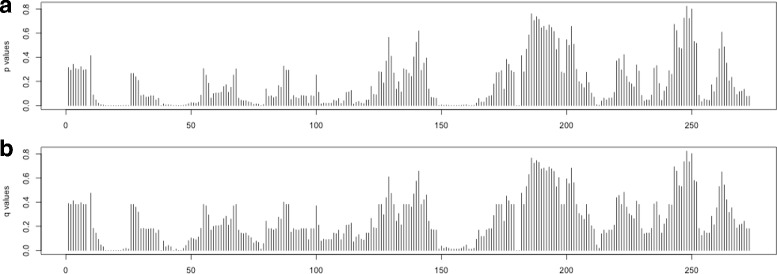
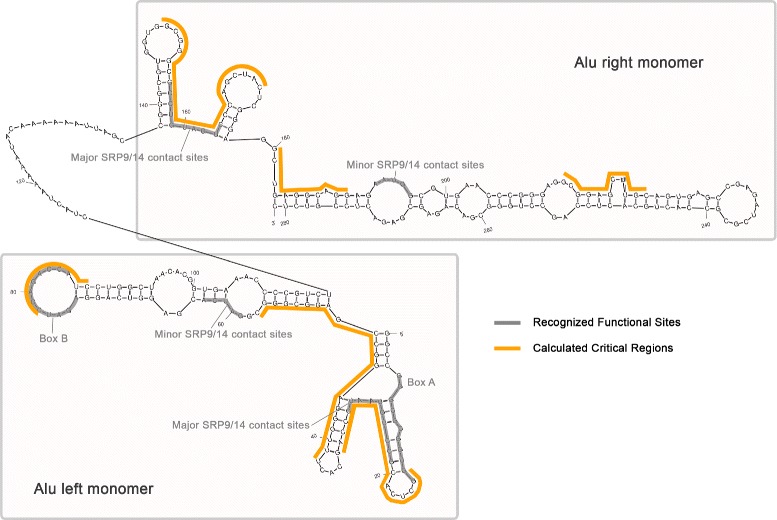
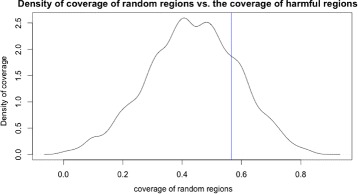
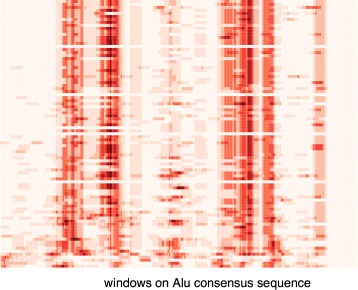
A method/simulation is created to computationally predict so-called harmful mutation regions in the consensus sequence of a TE; that is, mutations that occur in these regions decrease the transpositional activity dramatically. The methods are applied to the most active subfamily, AluY, to identify the harmful regions, and seven harmful regions are identified within the AluY consensus with q-values less than 0.05. A supplementary simulation also shows that the identified harmful regions covering the AluYa5 RNA functional regions are not occurring by chance. This method is then applied to two additional TE families: the Alu family and the L1 family, to computationally detect the harmful regions in these elements.
We use a computational method to identify a set of harmful mutation regions. Mutations within the identified harmful regions decrease the transpositional activity of active elements. The correlation between the mutations within these regions and the transpositional activity of TEs are shown to be statistically significant. Verifications are presented using the activity of AluY elements and the secondary structure of the AluYa5 RNA, providing evidence that the method is successfully identifying harmful mutation regions.
转座元件(TEs)是散布在 DNA 序列中的可移动或复制到基因组内新位置的序列。TEs 被认为促进了物种形成,其活动在人类疾病中起着重要作用。在人类基因组中,22 个 AluY 和 6 个 AluS TE 亚家族是最近最活跃的,它们的转座已被牵连到许多遗传性人类疾病和各种形式的癌症中。因此,了解它们的转座活性非常重要,确定影响它们转座活性的因素也非常有趣。最近,已经有一些工作基于序列变化来量化活性 Alu TE 的活性水平。有了这些活性数据,就可以基于突变的位置对 TE 活性进行分析。
创建了一种方法/模拟来计算 TE 一致性序列中所谓的有害突变区域;也就是说,这些区域发生的突变会极大地降低转座活性。该方法应用于最活跃的亚家族 AluY,以确定有害区域,并在 AluY 一致性中确定了七个 q 值小于 0.05 的有害区域。补充模拟还表明,覆盖 AluYa5 RNA 功能区域的识别出的有害区域并非偶然发生。然后将该方法应用于另外两个 TE 家族:Alu 家族和 L1 家族,以计算这些元件中的有害区域。
我们使用一种计算方法来识别一组有害突变区域。这些识别出的有害区域内的突变会降低活性元件的转座活性。这些区域内的突变与 TE 转座活性之间的相关性被证明具有统计学意义。使用 AluY 元素的活性和 AluYa5 RNA 的二级结构进行验证,提供了该方法成功识别有害突变区域的证据。